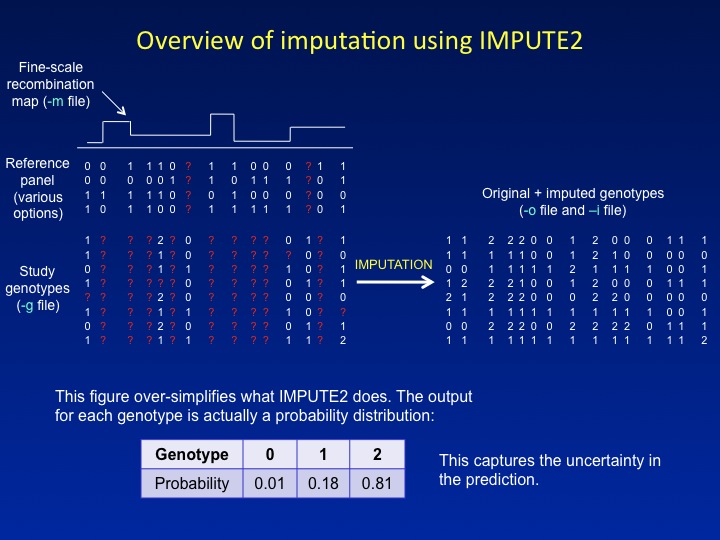
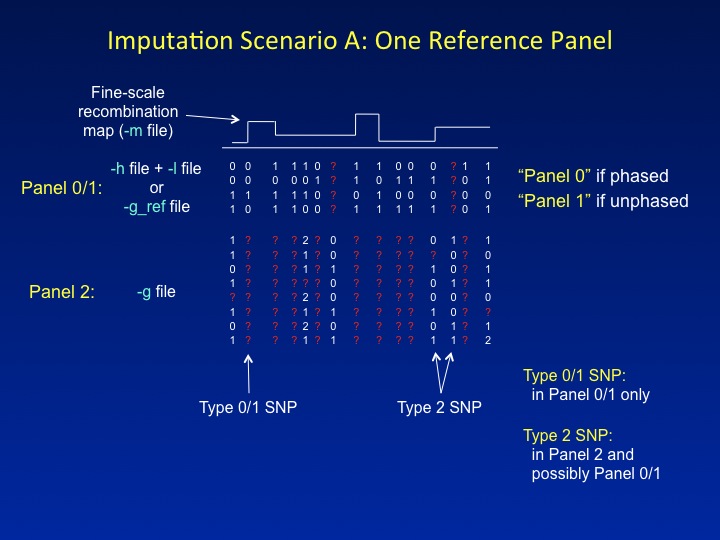
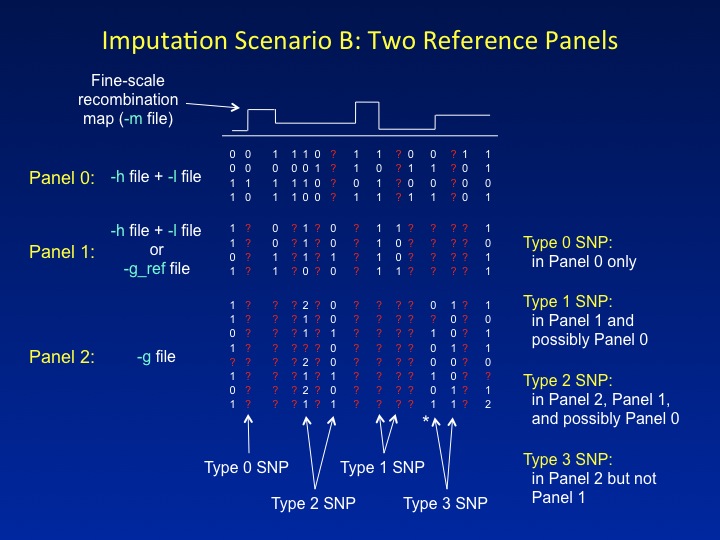
|
Flag
|
Default
|
Description
|
|
-o <file>
|
./test.impute2
|
Name of main output file. Follows the same
format
as the
-g file.
|
|
-i <file>
|
[-o]_info
|
Name of SNP-wise information file with one line per SNP and a single header line at the beginning;
versions of IMPUTE prior to v2.1.0 did not print the header.
This file always contains the following columns (header tags shown in parentheses):
1. SNP identifier from
-g file
(snp_id)
2. rsID
(rs_id)
3. base pair position
(position)
4. expected frequency of allele coded '1' in the
-o file
(exp_freq_a1)
5. measure of the observed statistical information associated with the allele frequency estimate
(info)
6. average certainty of best-guess genotypes
(certainty)
7. internal "type" assigned to SNP
(type)
-- column did not exist prior to v2.1.0
Depending on the command-line options invoked, there may also be columns labeled
info_typeX,
concord_typeX,
and
r2_typeX,
where
X
takes values in
{0,1,2}.
For SNPs that have genotypes in the
-g file,
concord_typeX
is the concordance between the input genotypes (after applying the
-call_thresh)
and the best-guess imputed genotypes obtained by masking the input genotypes one SNP at a time
and pretending the SNP is of type
X;
similarly,
r2_typeX
is the squared correlation between input and imputed genotypes.
The
info_typeX
column is the same information metric used in column 5, but here is it applied to genotypes that
have been imputed from pseudo-type
X
SNPs in the leave-one-out masking experiment.
These columns did not exist prior to v2.1.0. They are useful
for post-hoc quality control, as we explain in the section on
QC and troubleshooting.
|
|
-r <file>
|
[-o]_summary
|
Name of file that records a summary of the screen output.
|
|
-w <file>
|
[-o]_warnings
|
Name of file that records warnings generated by IMPUTE2.
|
|
-os <int> <int> ...
|
0 1 2 3
|
"Output SNPs": specifies the SNP types that will be printed to the output file (SNP labeling
is discussed in the Overview). By default, all imputed and genotyped
SNPs are included in the output, i.e.,
"-os 0 1 2 3".
|
|
-o_gz
|
|
Specifies that the main output file should be compressed by the
gzip utility; this also applies
to some non-standard output files that can become large.
|
|
-outdp <int>
|
3
|
Specifies the number of decimal places to use for reporting genotype probabilities
in the main output file.
|
|
-no_snp_qc_info
|
|
Suppresses printing of
info_typeX,
concord_typeX,
and
r2_typeX
columns in
-i file.
|
|
-no_sample_qc_info
|
|
Suppresses printing of per-sample quality control metrics file. The default is to print a file named
"[-i]_by_sample".
|
|
-phase
|
|
IMPUTE2 always implicitly phases the study dataset
(-g file),
and this flag tells the program to print the best-guess haplotypes that result
from the phasing process. In addition to the standard imputation output file, the program
also prints a separate haplotypes file named
"[-o]_haps".
This file contains two columns (haplotypes) per individual, in the same order they appear in the main output.
In addition to this "best-guess" haplotypes file, the program also prints the
certainty that each successive pair of heterozygous SNPs is correctly phased. These
certainties occur in a file named
"[-o]_haps_confidence".
This file contains one line per individual and one column per SNP in the phased
haplotypes file. Homozygotes are represented by * characters and heterozygotes
are represented by numbers between 0.5 and 1.0; this is the estimated probability
that the phasing between the current heterozygote and the previous heterozygote (to the
left) is correct. By convention, the leftmost heterozygous SNP in each individual is
assigned a phasing certainty of 1.0.
As shown in the examples section, it is possible to
use the
-phase
option to produce haplotypes without the use of a reference panel; i.e., to perform a
classical phasing analysis.
|
|
-pgs
|
|
"Predict Genotyped SNPs": Tells the program to replace the input genotypes from the
-g file
with imputed genotypes at Type 2 SNPs in the output file.
|
|
-pgs_miss
|
|
Unlike
-pgs,
this option tells the program to replace only missing genotypes with imputed genotypes.
That is, any input genotype whose maximum probability exceeds the
-call_thresh
will simply be reprinted in the output file, whereas input genotypes that fall below the
calling threshold will be imputed in the output.
WARNING:
This is an appealing option that promises to simply "fill in" sporadically missing genotypes
in your input data. However, we think that following this procedure and then testing the SNPs
for association could cause subtle problems. We are investigating
these issues, but in the meantime we suggest that you only use this option with great
caution; using it naively may lead to bad results, and you do so at your own risk.
|