Overview (top)
The
figure below provides a schematic overview of what IMPUTE2
does. In short, it uses a fine-scale recombination map and a densely
genotyped reference panel to "fill in" missing genotypes in a study
dataset, which might consist of cases and controls typed on a
commercial SNP chip. By estimating the genotypes of SNPs that were not
in the original study data, imputation allows a much larger set of SNPs
to be tested for association. This can increase both the power to
detect association signals and the signal resolution near a causal
variant. 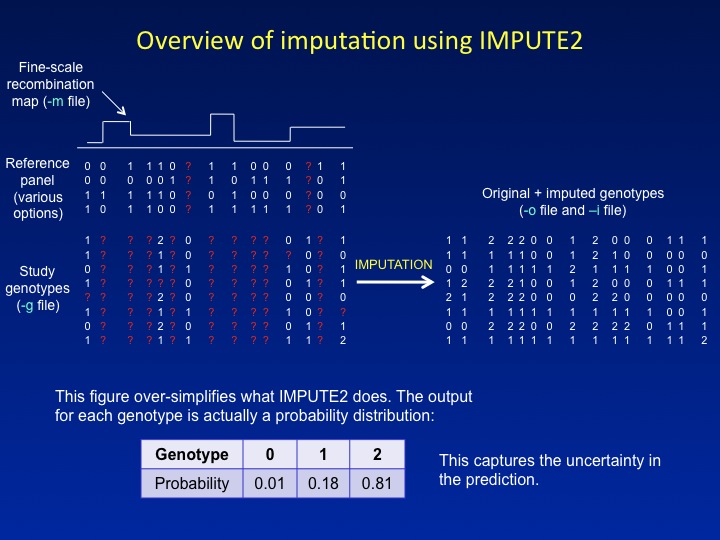
Imputation scenarios and program nomenclature
The next two figures illustrate the common imputation scenarios that IMPUTE2 is designed to handle. These figures introduce the nomenclature used by the program to label panels and SNPs, including "special" SNPs that do not fit into the standard imputation framework.SCENARIO A: ONE REFERENCE PANEL
This is the imputation setup that most people are familiar with: a reference panel containing a dense set of SNPs is used to impute missing genotypes in a study dataset that has been typed at a sparser set of SNPs. IMPUTE2 refers to the reference data as Panel 0 or Panel 1 (for phased and unphased reference panels, respectively) and to the study data as Panel 2. These labels serve as a convenient shorthand in the program's screen output.
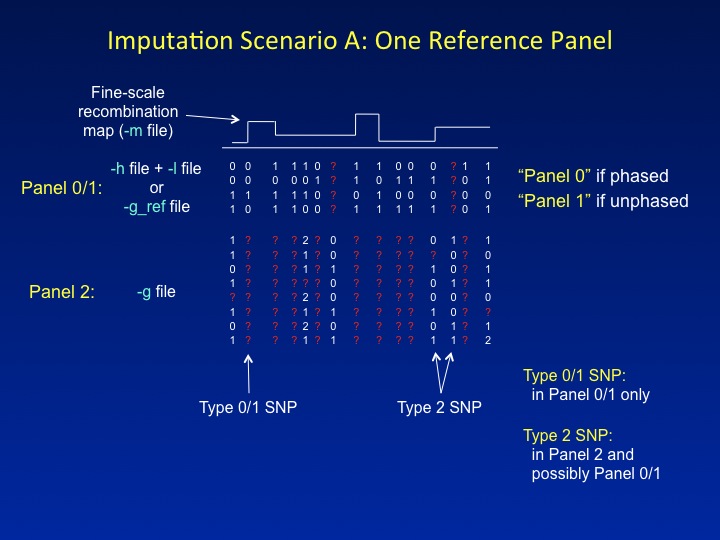
IMPUTE2 labels SNPs by the panels in which they have been genotyped. Each label denotes a specific functional role. In the figure above, SNPs that have data only in the reference panel are labeled Type 0 or Type 1 (for phased and unphased reference panels, respectively), whereas SNPs that have genotypes in the study dataset are labeled Type 2. Type 2 SNPs dictate which reference panel haplotypes should be "copied" for each individual; then, the reference panel alleles at Type 0/1 SNPs are used to fill in that individual's missing genotypes.
There is one novelty in the way that IMPUTE2 treats Scenario A. In the figure, one of the SNPs that is labeled as Type 2 has data in Panel 2 but not in the reference panel. Most imputation methods ignore these kinds of SNPs since they are hard to model. For example, IMPUTE v1 labels these as Type 3 SNPs, and it does not impute them or use them to inform the inference. By contrast, IMPUTE2 uses a novel approach to model the missing reference panel alleles, thereby allowing it to gain information from the study genotypes at such SNPs. This feature highlights one of the guiding principles of IMPUTE2: to increase imputation accuracy by using as much of the information in the data as possible.
SCENARIO B: TWO REFERENCE PANELS
Another novel feature of IMPUTE2 is the ability to combine two reference panels containing different sets of SNPs in a single imputation analysis. In the figure below, the first reference panel is called Panel 0, the second reference panel is called Panel 1, and the study dataset is called Panel 2. It is common for each successive panel (0,1,2) to be genotyped at a subset of the SNPs in the previous panel. For example, Panel 0 might comprise haplotypes from the 1,000 Genomes Project, which captures nearly all common SNPs in the genome; Panel 1 might comprise haplotypes from HapMap Phase 3, which surveys a subset of common SNPs; and Panel 2 might be a set of cases and controls genotyped on a commercial SNP chip. IMPUTE2 assumes that the sets of SNPs in
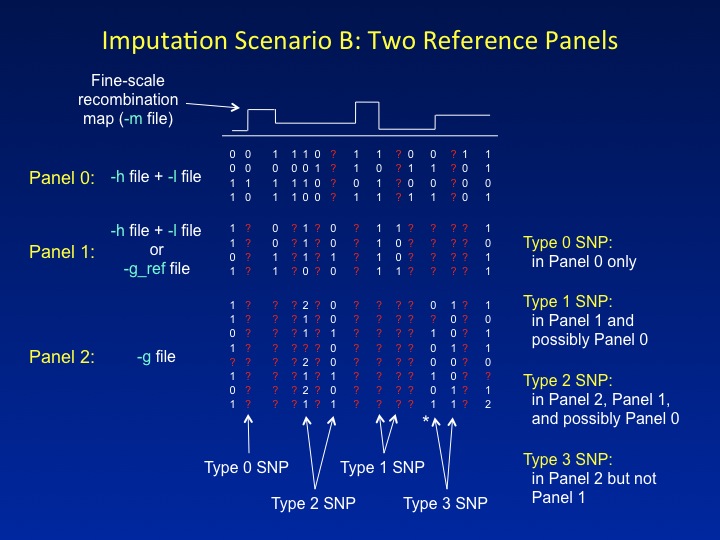
In imputation Scenario B, SNPs are labeled as follows:
- Type 0 SNPs have data in Panel 0
only and are used for imputation.
- Type 1 SNPs have data in Panel 1
and are used for imputation. They may or may not have data in Panel 0;
if
not,
IMPUTE2 will simulate the Panel 0 alleles
with its hole-filling function.
- Type 2 SNPs have data in Panel 2
and Panel 1, and are used to determine which reference
panel haplotypes will be copied. They may or may not have data in Panel 0;
if
not,
IMPUTE2 will simulate the Panel 0 alleles
with its hole-filling function.
- Type 3 SNPs have data in Panel 2 only*. These SNPs do not fit easily into the imputation model, so their genotypes cannot inform the inference.