MVNcall
MVNCALL is a program
developed for genotype calling and phasing using low-coverage
next-generation sequencing reads information. The
software assumes a study design where the samples are
sequenced and also genotyped in a microarray. The approach
assumes that the microarray genotypes have been phased using a
haplotype estimation program. We recommend SHAPEIT for this
purpose. The phased haplotypes then serve as a haplotype scaffold in MVNCALL.
Then, each polymorphic site is phased one at a time onto the
haplotype scaffold.
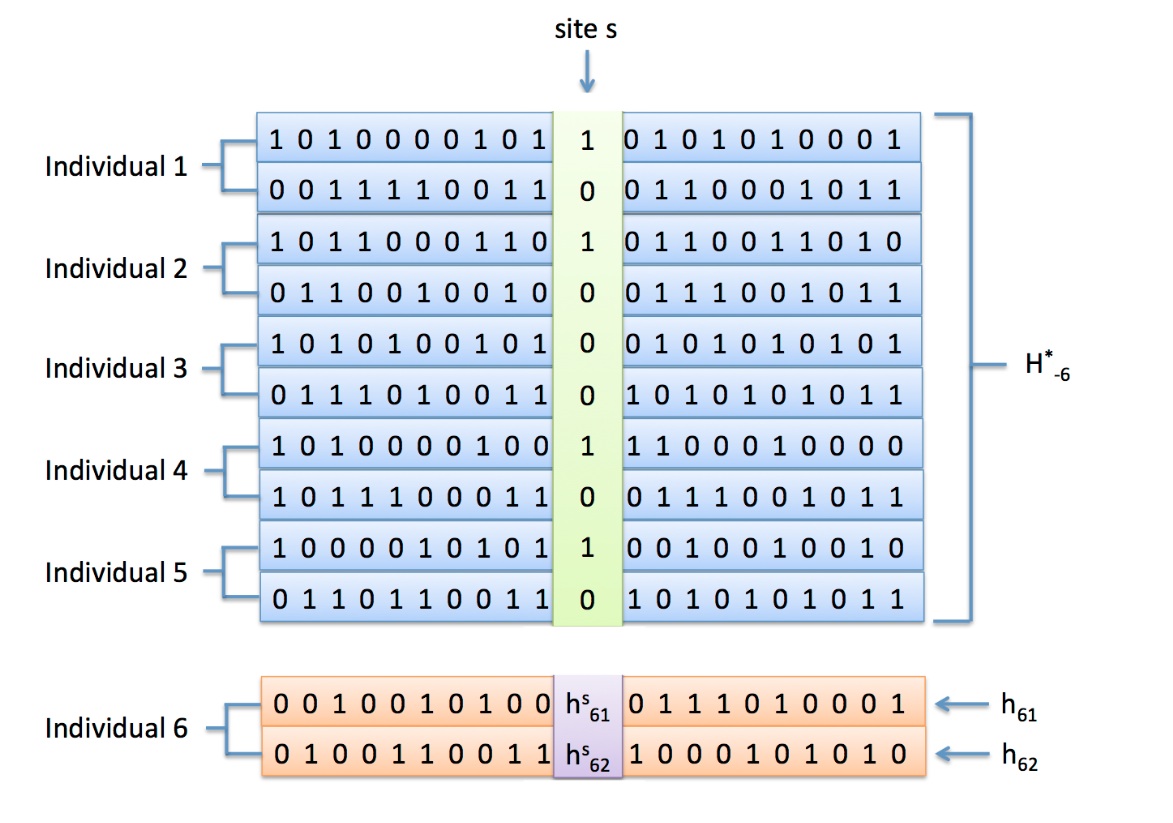
In the figure below illustrates how MVNcall
works. The method implements an MCMC algorithm in which
unobserved genotypes are iteratively updated. The figures
illustrates how a site s is phased onto the haplotype
scaffold. At each iteration, the posterior genotypes of an
individual (in purple) are estimated given the (i) haplotype
scaffold (in blue), (ii) the genotypes at the site at all
other individuals (in green), and (iii) the flanking
haplotypes of the individual in a given iteration (in orange).
The approach is described in our paper
A. Menelaou and J.
Marchini (2013)
Genotype calling and phasing using next-generation sequencing
reads and a haplotype scaffold. Bioinformatics 29(1):84-91[Link]
MVNCALL
has the following properties:
- It analyses one site at a time
- It is highly parallelizable
- It requires low RAM usage.
- It can leverage duo/trio information when available
- It can analyse multi-allelic
variants
What's New?
First software release (30 December 2013)
We have just released the first version MVNcall v1.0
Download MVNcall
MVNcall is freely available for academic use. To see rules for non-academic use, please read the LICENCE file, which is included with each software download.
Pre-compiled MVNcall binaries and example files can be downloaded from the links below. For Linux machines, the dynamic binaries are smaller but may not work on some machines due to gcc library compatibility issues; if the dynamic version doesn't work for you, please try the static version. If you have any problems getting the program to work on your machine or would like to request an executable for a platform not shown here, please send a message to our mail list.
The latest software release is v1.0. We support only the most recent version.
| Platform | File |
|---|---|
| Linux (x86_64) Dynamic Executable | mvncall_v1.0_x86_64_dynamic.tgz |
To unpack the files on a Linux computer, use a command like this:
tar -zxvf mvncall_v1.0_i686.tgz
(Other file decompression programs are available for non-Linux
computers.) This will create a directory of the same name as the
downloaded file, minus the '.tgz' suffix. Inside this directory
you will find an executable called mvncall,
a LICENCE file, and an Example/ directory that contains example
data files. We show how to perform various kinds of analyses
with the example files here.
Examples
This section provides some example commands that illustrate typical applications of MVNcall. All of the data files used in these commands are included in the Example/ directory that comes with the software download. You should run the commands from the main download directory (i.e., the one that contains the mvncall executable). Detailed explanations are provided at each link below.
| Run type | Description |
|---|---|
| Imputation with default parameters | Basic scenario in which most people will use MVNcall. |
| Imputation with model averaging option | As above, but with model averaging which averages over two models. |
| Imputation with duo/trio related samples | As above, but with related samples. |
How to use example commands
All of the data files in the example commands below are
included in the example/ directory that comes with the MVNcall
software download. You should run the command from the main
download directory, which is the one that contains the mvncall executable. For example, if you
just downloaded a software package named mvncall_v1.0_MacOSX.intel.tgz and
unpacked it according to the directions here,
you can reach the appropriate directory by typing "
Once you have found the right directory, you should be able to run the example command by entering it into a Unix-style terminal window. Depending on the settings of your computer, this may be as simple as highlighting the command text in your web browser, using the browser's Copy command, and then using the Paste command in your terminal window. (You may then need to hit Enter to start the run.)
Note that most lines in the example command end with the '\' character. This is not actually part of the command; it is just a shorthand notation that means "keep reading the next line as part of a single command." We use this notation to split the command over multiple lines so it is easier to read. This is a valid way to enter commands in a Unix-style terminal window, but it would be equivalent to put all of the arguments on a single line, separated by spaces.
You do not have to run MVNcall exactly as in the example. Some of the arguments shown here are optional, and there are many other options that could be added to modify the behavior of the program. For a full list of available options, see here.
Most of the examples below include the string "
Imputation with DEFAULT PARAMETERS
This is the default scenario using the default parameters (for
--iteration, --burn-in, --lambda,
--numsnps).
The following command shows how to run this kind of analysis with MVNcall, using the example data that come with the program download:
./mvncall \
Comments
- Here we input the --sample-file
which corresponds to the order of the haplotypes in the --scaffold-file. The --glfs file is in vcf
format. Care should be taken that the ids in the --sample-file
match the ids in the --glfs file.
- The --int specifies the interval of sites to be imputed. Only sites that are present in the --glfs sites will be imputed. In the case where a site is present in the --scaffold-file, no imputation is performed, and the site is kept the same as in the --scaffold-file.
- The --o provides the phased genotypes at the sites analysed in a vcf format.
- The default values for the other parameters are used for the
imputation of the Phase
1 of the 1000 Genomes Project. For different study
designs those parameters might need to be modified.
Imputation with MODEL AVERAGING OPTION
The number of conditioning haplotypes (--k)
is fixed to a single value. In order to run MVNcall with
two values of --k,
one specified by the --k flag and the other with all
available haplotypes, the model-averaging option can be used.
The two models are ran simultaneously and the averaged
posteriors are used to infer the phased genotypes. This
procedure seemed to help the imputation of the Phase 1 1000
Genomes data.
The following commands show how to run this kind of analysis with MVNcall, using the example data that come with the program download:
./mvncall \
--k 100 \
Comments
- The --model-avg option runs the model with two values of --k (100 and all haplotypes). For low coverage sequencing data, the model averaging option was preferred since it provided lower genotype discordance rates. The computational time is however increased. We recommend you first test the --model-avg option in a small region and compare the genotype discordance with the model ran on a single value of --k before applying it to larger regions.
- Instead of choosing an interval through the --int flag
to impute with MVNcall, a list of sites can also be inputted
where the physical positions of the sites analysed are
inputted, one in each row. Note that no chromosome is required
in the list, since MVNcall assumes that a single
chromosome is inputted.
Imputation with DUo/TRIO RELATED SAMPLES
This scenario assumes that the samples are related (either
duos -parent,offspring-, or trios -both parents,offspring-). We
highly recommend to use the known relationships of the samples,
if available, since this has shown to significantly help both
the imputation and phasing of the samples.
The following command shows how to run this kind of analysis with MVNcall, using the example data that come with the program download:
./mvncall \
Comments
- The --ped-file should follow this format. Make sure that the ids in the ped file agree with the ped files in --sample-file.
- The transmitted haplotypes in the --scaffold-file should be:
father transmits the first haplotype to the first haplotype of
the offspring, and the mother transmits the first haplotype to
the second haplotype of the offspring. This
is crucial for running the pedigree related model.
Program Options
| Flag | Default | Description |
|---|---|---|
|
--sample-file
<file> REQUIRED |
none | File containing the IDs of the samples in the haplotype scaffold. There should be no header in the file. The order of the IDs in the the sample file needs to agree with the order of haplotypes in the scaffold file. An example of a sample file can be found here |
|
--scaffold-file <file> REQUIRED |
none | File containing the haplotypes of the samples as
specified in the sample file. Each row corresponds to a
SNP and each column to a haplotype. The first five columns
of the file give information regarding the chromosome, the
position, and the alleles. For more details see here. |
| --glfs
<file> REQUIRED |
none | The glfs file is a vcf
format file that contains the genotype likelihoods
of the sites to be analysed on the same set of samples
that are in the sample file. The genotype likelihoods can
be in GL or PL format. When multiple variants have the same
physical position, the output will contain
pseudo-positions with +1 from the input position. The option supports .gz format. |
| --ped-file
<file> |
none |
The ped-file contains the duo/trio
relationships between the samples studied. The file must
follow this
format. When a ped-file is not given, then the
individuals are assumed to be unrelated. For more details
on the format see here. |
| --o <file>
REQUIRED |
none |
The output file contains the phased
genotypes in a vcf format. For more details see here |
| --mp <file> |
none |
The file contains the marginal posterior
probabilities of the phased genotypes. For
bi-allelic sites, the order is RR-AA-AR-RA where
R=reference allele and A=alternative allele. For more than
2 alleles, the order follows the same convention as used
in vcf format. At each row the position, the reference and
alternative allele are given in the first three columns
and the following columns correspond to the marginal
probabilities of the individuals at the site. An example
of the output file is given here |
| --int
<lower> <upper> |
none |
Genomic interval to use for inference, as
specified by <lower> and <upper>
boundaries in base pair position. The option is useful for
splitting the chromosome in multiple regions and run them
parallel. |
| --list
<file> |
none |
Instead of using a genomic interval, a list
of sites with their genomic positions can be given. Only
the positions in the list will be imputed. An example of a
list file is given here |
| --numsnps
<int> |
50 |
The number of flanking sites that is going
to be used for inference at either site of the site of
interest. |
| --lambda
<double> |
0.06 |
The penalty parameter added on the diagonal
of the variance-covariance matrix. The higher the value of
lambda, the more weight is given to the GLs and less to
the LD structure to calculate the posterior probabilities
of genotypes. The default value is suggested for
low/medium coverage data. |
| --k <int> |
all |
The number of conditioning haplotypes to be
used for inference. Each haplotype in a given
individual, conditions on k haplotypes, which do not need
to be the same. Perfect matching is used for choosing the
closest haplotypes. We recommend to use different
values of k before you decide which value to use. For the
1000 Genomes Phase 1 Project and the Genome of the
Netherlands, we use k=100 |
| --model-avg |
none |
Model average flag will run MVNcall twice:
once with the value of k as indicated in --k,
and another one with k equal to all haplotypes. |
| --iteration
<int> |
100 |
Total number of MCMC iterations to perform,
including burn-in. Increasing the number of
iterations may improve accuracy, depending on the number
of samples and the depth of coverage. |
| --burn-in
<int> |
10 |
Number of MCMC iterations to discard as
burn-in. |
File formats
Examples of file formats required as inputs in MVNcall are
provided.
- Sample
file (
--sample-file ): The order of the samples in the haplotype files are required in all analyses. The sample file contains the IDs of the samples, one in each row. No header file is required.
IND1
IND2
IND3
IND4
- Scaffold
file (
-scaffold-file ): The study design assumed by MVNcall includes a phased haplotype scaffold. A haplotype scaffold is built by phasing microarray genotypes on the same set of samples that are sequenced. We recommend SHAPEIT2 for building the haplotype scaffold. The output of SHAPEIT2 includes the HAPS and SAMPLE file formats for haplotypes. The scaffold file has the same format as the SHAPEIT2 HAPS files.
1 SNP2 456 G C 0 0 1 0 1 0
1 SNP3 789 T A 0 1 1 1 0 1
- Chromosome number <integer>
- SNP ID <string>
- SNP Position <integer>
- First allele <string>
- Second allele <string>
- The subsequent column pair (6,7), (8,9), (10,11), (12,13) corresponds to the alleles carried at that SNP by each haplotype of the four individuals.
- Ped file (
--ped-file ): This is a space delimited file which includes at least five columns. The five columns correspond to:
- Family ID <string>
- Individual ID <string>
- Father ID <string>
- Mother ID <string>
- Sex <integer>
- Phenotype <float>
FAM1 IND1 0 0 1 0
FAM2 IND2 0 0 2 0
FAM3 IND3 IND1 IND2 2 0
- Output file
(
-o ): The output file is in vcf format. The phased genotypes are given using the vcf notation, i.e. 0|0, 0|1, 1|0, 1|1. - Posterior probabilities file (--mp): The output file
gives the posterior probabilities of the phased
genotypes. For example:
123 A T 1 0 0 0 1 0 0 0 1 0 0 0
456 G C 1 0 0 0 0 1 0 0 1 0 0 0
789 T A 0 0 0 1 0 1 0 0 0 0 1 0
_____________________________________________________________________________________________________________________________________
References
A. Menelaou and J.
Marchini (2013)
Genotype calling and phasing using next-generation sequencing
reads and a haplotype scaffold. Bioinformatics 29(1):84-91[Link]Contributors
The following people developed the methodology and software for MVNcall:
Androniki Menelaou, Jonathan Marchini
Mail List
If you have a question about MVNcall, please send a message to our mailing list:
http://www.jiscmail.ac.uk/OXSTATGEN
You will need to subscribe to the mailing list to post a question. The list has low but steady traffic, so you may want to redirect the messages to a dedicated e-mail folder if you don't want them all landing in your inbox.
IMPORTANT: If you are having a problem with the software, please include the following details in your e-mail; otherwise, we may not be able to diagnose the problem.
- The version number of MVNcall and the type of
computer you are using to run it—e.g.,
"MVNcall v1.0 on Mac OSX 10.6". - Any log files and/or screen output from the program;
- For difficult problems like memory access errors (e.g., "segmentation faults"), we may need you to send data files that show the problem. These files should ideally be small, and we can provide suggestions if you are not allowed to share your actual data.