| Home | X Chromosome Imputation |
| Overview |
Options |
| Contributors |
FAQ |
| Download |
References |
| Running IMPUTE | Contact Information |
| Using IMPUTE with the HapMap Data | Version History |
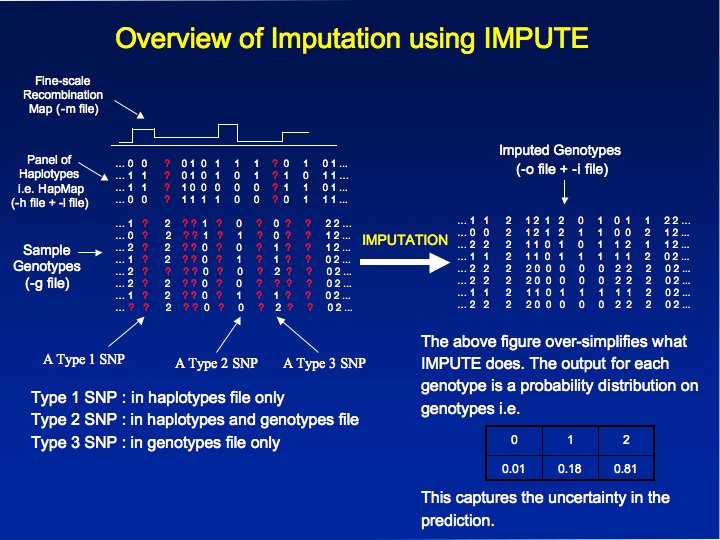
IMPUTE is a
program for
imputing unobserved genotypes in genome-wide
case-control studies based on a set of known haplotypes (like the
HapMap Phase II haplotypes [2]). The
program
is designed to work seamlessly with the output of both
the genotype calling program CHIAMO [1] and HAPGEN and
produce output that can be analyzed using the program SNPTEST [2]. This program was
used to carry out genotype imputation as part of the analysis of the 7
genome-wide association studies analyzed by the Wellcome Trust
Case-Control Consortium (WTCCC) [3].
|
|
| Platform |
File |
| Linux
(x86_64) Static Executable |
impute_v0.5.0_x86_64_static.tgz |
| Linux
(x86_64) Static Executable (SuSE 9.3) |
impute_v0.5.0_SuSE9.3_x86_64_static.tgz |
| Linux
(x86_64) Dynamic Executable |
impute_v0.5.0_x86_64_dynamic.tgz |
| Linux
(i386) Static Executable |
impute_v0.5.0_i386_static.tgz |
| Linux
(i386) Dynamic Executable |
impute_v0.5.0_i386_dynamic.tgz |
| Mac OS X 10.5.01 Tiger (Intel) | impute_v0.5.0_MacOSX_10.4_Intel.tgz |
| Mac
OS X 10.5.1 Leopard (Intel) |
impute_v0.5.0_MacOSX_10.5_Intel.tgz |
| Mac OS X (PowerPC) | impute_v0.5.0_MacOSX_PowerPC.tgz |
| Solaris
5.8 (Sun SPARC) |
impute_v0.5.0_Solaris5.8_SPARC.tgz |
| Solaris
5.10 (AMD Opteron) |
impute_v0.5.0_Solaris5.10_Opteron.tgz |
| SLES 10 (Intel Itanium2) | impute_v0.5.0_Itanium2_SLES10.tgz |
| Windows
MS-DOS (Intel) |
impute_v0.5.0_Windows_Intel.tgz |
| tar zxvf impute_vX.X.X_i386.tgz |
| ./impute -h
example/haplo.txt -l example/legend.txt -g example/geno.txt -m
example/map.txt -s example/strand.txt -Ne 11400 -int 62000000 63000000 |
| bash$
./impute -h example/haplo.txt -l example/legend.txt -g example/geno.txt
-m example/map.txt -s example/strand.txt -Ne 11400 -int 62000000
63000000 IMPUTE v0.5.0 ============= Copyright 2006 Jonathan Marchini Please see the LICENCE file included with this program for conditions of use. haplotypes file : example/haplo.txt legend file : example/legend.txt genotypes file : example/geno.txt map file : example/map.txt <---- list of input and output files strand file : example/strand.txt output file : ./out info file : ./info imputation interval : [62000000,63000000] <---- specifies the region of imputation from -int option reading genetic map...done reading haplotypes # ind = 120 # snps read in = 1129 reading genotypes # ind = 50 # SNPs with genotypes read in = 250 reading strand file # SNPs in strand file = 250 # SNPs in imputed region that have had strand assigned = 250 Summary : 122 SNPs in left-hand buffer region 223 SNPs in right-hand buffer region 662 type 1 SNPs will be in output file (type 1 = SNP in haplotype file only) 141 type 2 SNPs will be in output file (type 2 = SNP in haplotype file and genotype file) 27 type 3 SNPs will be in output file (type 3 = SNP in genotype file only) 830 SNPs will be in output file in total 1175 SNPs in total -using strand file to orientate strand --flipped strand at 103 genotyped SNPs out of a total of 204 <---- details of strand alignment -aligning allele labels of haplotypes and genotypes -removing non-aligned genotyped SNPs --removing 0 genotyped SNPs out of a total of 204 setting weights...done setting storage space...done setting mutation matrices...done setting switch rates...done Estimated RAM required is 74.115Mb n_hap : 120 n_gen : 50 nind : 50 interval : [62000000, 63000000] buffer : 250 <--- this is the buffer region (in kb) used on each end of the region to avoid edge effects Ne : 11400 <--- this is the Ne value used in the model call_thresh : 0.900 <--- this is the threshold used to call genotypes from the input genotype file theta : 0.18655 model : 4 predicting individual [50/50] [forward sweep] [backward sweep] [predict] Breakdown of impution accuracy at SNPs with genotypes in the input file This assessment only uses genotypes in input file that are called above threshold of 0.90 There are 7024 such genotypes in total For each of these genotypes the maximum imputed genotype calls are distributed as follows Interval #Genotypes %Concordance Interval %Called %Concordance [0.0-0.1] 0 0.0 [ >= 0.0] 100.0 95.9 [0.1-0.2] 0 0.0 [ >= 0.1] 100.0 95.9 [0.2-0.3] 0 0.0 [ >= 0.2] 100.0 95.9 [0.3-0.4] 0 0.0 [ >= 0.3] 100.0 95.9 [0.4-0.5] 32 40.6 [ >= 0.4] 100.0 95.9 imputation accuracy [0.5-0.6] 175 51.4 [ >= 0.5] 99.5 96.1 <--- For genotypes in the input file [0.6-0.7] 155 65.8 [ >= 0.6] 97.1 97.3 this says that using a calling [0.7-0.8] 163 77.3 [ >= 0.7] 94.8 98.0 threshold of 0.5 99.5% of [0.8-0.9] 305 82.3 [ >= 0.8] 92.5 98.5 imputed genotypes would be [0.9-1.0] 6194 99.3 [ >= 0.9] 88.2 99.3 called and 96.1% of those are concordant/correct. finito <--- this says 'I am finished' in Italian |
| ./impute -h
example/haplo.txt -l example/legend.txt -g example/geno.txt -m
example/map.txt -fix_strand -Ne 11400 -int 62000000 63000000 |
| ./impute -h
example/haplo.txt -l example/legend.txt -g example/geno.txt -m
example/map.txt -s example/strand.txt -Ne 11400 -int 62000000 63000000
-pgs -os 2 |
| ./impute -h
example/haplo.txt -l example/legend.txt -g example/geno.txt -m
example/map.txt -s example/strand.txt -Ne 11400 -int 62000000 63000000
-exclude_snps example/exclude.txt -impute_excluded |
| Polymorphic
files - these files
contain SNPs polymorphic in each panel respectively i.e. the CEU haplotypes only contain data at SNPs that are polymorphic in the CEU panel. The files contain the haplotypes and associated legend files. |
[CEU] [YRI] |
| Recombination
rate files (nb. these are the same as the rel#22 rates) |
[CEU] [YRI] [COMBINED] |
| Strand files | Affy500k [These were
constructed using these Affymetrix annotation files - Nsp
Sty]
Affy6.0 [These files were created using this Affymetrix annotation file - LINK] |
| Polymorphic
files - these files
contain SNPs polymorphic in each panel respectively i.e. the CEU haplotypes only contain data at SNPs that are polymorphic in the CEU panel. The files contain the haplotypes and associated legend files. |
[CEU] [YRI] [JPT+CHB] |
|||||||
| Consensus
files - these files
contain SNPs that occur in all 3 of the HapMap panels. There are also files for all combinations of the panels, which are useful for imputation of admixed individuals. |
|
|||||||
| Recombination rate files | [CEU] [YRI] [COMBINED] | |||||||
| Strand files | Affy500k [These were
constructed using these Affymetrix annotation files - Nsp
Sty]
Affy6.0 [These files were created using this Affymetrix annotation file - LINK] |
| Polymorphic
files - these files
contain SNPs polymorphic in each panel respectively i.e. the CEU haplotypes only contain data at SNPs that are polymorphic in the CEU panel. The files contain the haplotypes and associated legend files. |
[CEU] [YRI] [JPT+CHB] |
| Recombination rate files | [CEU] [YRI] [JPT+CHB] [COMBINED] |
| Strand files | Affy500k [These files were constructed using these Affymetrix annotation files - Nsp Sty] |
| ./impute -chrX -h
chrX_files/genotypes_chrX_CEU_r21_nr_fwd_non-par_phased_by_snp_no_mono
-l chrX_files/genotypes_chrX_CEU_r21_nr_fwd_non-par_legend.txt -m
chrX_files/genetic_map_chrX_non-par.txt -s
chrX_files/Affy500k_chrX_non-par.strand -g chrX_files/chrX.example.gen
-sample chrX_files/chrX.example.sample -Ne 11400 -int 4000000
4100000 |
| Flags |
Required/Optional |
Default | Description |
| -h
<file> |
Required |
File
containing a set of
known
haplotypes for the region of interest. The alleles of the haplotypes
should be coded as 0 and 1. The format of this input file is
one line per SNP and one column per haplotype. |
|
| -l
<file> |
Required | Legend
file for haplotypes
file which give rs ID, position and the alleles that are coded as 0 and
1 in the haplotypes file. The alleles should be taken from A, C, G and
T. Note that this file needs a header line (see the example file
legend.txt for details) |
|
| -g
<file> |
Required | File containing a set of
genotypes for the set if individuals. The file format is described in
detail on the FILE
FORMAT WEBPAGE. The file format is the same as the output
format from our genotype calling program CHIAMO.
NOTE 1 : The SNPs MUST appear in base-pair position order (lowest to highest) i.e. the 3rd column of this file must be sorted. NOTE 2 : Base-pair positions of SNPs must use the same genome build as that used in the haplotype file. |
|
| -g_gz |
Optional |
Specify that the genotype file
is gzipped. |
|
| -m
<file> |
Required | Fine-scale recombination map covering the region at which impution is required. There is one line for each position on the map. The first column contains the base pair position, the 2nd column contains the recombination rate in cM/Mb to the next point on the map and the 3rd column contains the recombination map position in cM.Note that this file needs a header line (see the example file map.txt for details) | |
| -Ne
<int> |
Required | Sets effective population size that scales the fine-scale recombination map for the given population. For example, -Ne 11000 sets the effective population size to 11000. For autosomal chromosomes, we highly recommend the values 11418 for CEPH, 17469 for Yoruban and 14269 for Chinese Japanese populations. | |
| -int
<lower> <upper> |
Required |
Lower are Upper boudaries (in base pair position) of the region in which imputation should be carried out. | |
| -s
<file> |
Optional | File
listing the strand
orientation of the SNPs in the genotype file relative to the
orientation of the alleles in the haplotypes file. This is file is
required if the orientation of alleles at SNPs in the haplotype and
genotype files does not match up. The file should contain a line for
each SNP in the genotype file with two entries (i) the base-position of
the SNPs, and (b)
the strand (+ or -) of the alleles in the genotype file. SNPs do
not have to be in the same order as in the genotypes file and the file
can include SNPs that are not in the genotypes file i.e. if the
genotypes file has had some SNPs filtered out. Take a look at the
example files
for an illustration of the required format. NOTE : It is critical that the alleles used to code genotypes in the haplotype file and the genotype file match up. If not, then the quality of imputation may decrease substantially. Great care should be taken in constructing a strand file for your data. NOTE : see the -fix_strand and -no_remove options below which control the internal strand alignment functions. |
|
| -fix_strand |
Optional |
This flag invokes an internal strand alignment at SNPs that occur in both the genotypes and haplotypes files. It is based on the allele labels (at non A/T and G/C SNPs), and discorandant allele frequencies (at A/T and G/C SNPs ). | |
| -no_remove |
Optional |
This flag turns off the default
removal of all SNPs in the genotype file that are not aligned.
The removal of SNPs is carried out after any specified strand file has
been applied and after the checks described in the previous option have
been applied. |
|
| -o
<file> |
Optional |
./out |
Name of main output file that will contain the imputed genotypes. The files has one line per SNP and has exactly the same format as the genotypes file format. NB the program will estimate probabilities for all genotypes including those that are known in the genotypes file (this allows an asssesment of genotyping errors and imputation of missing data at these SNPs) |
| -o_gz |
Optional |
Specify that the output file
should be gzipped. |
|
| -i
<file> |
Optional | ./info |
Name of the file that
information measures that describe theaccuracy of imputation at each
SNP. This
file contains one line per SNP that contains SNP ID, rs ID,
position, expected allele frequency of the SNP, a measure of the
observed statistical information associated with the estimate of the
allele frequency and an alternative confidence score for the SNP
(calculated as the average of
the maximum posterior probabilities of the imputed genotypes). The
information measure and the confidence score will be 1 if the SNP is
imputed with hign confidence. Both measures decrease towards 0 as
imputation confidence decreases. |
| -r
<file> |
Optional |
./summary |
Specify file where a copy of the
screen output is written. |
| -buffer
<int> |
Optional |
250 |
To avoid edge effects in the imputation the program includes genotypes either side of the interval specified by the the -int flag. This option specifies the length of the buffer region (in kb) at each end of the interval. |
| -call_thresh
<double> |
Optional |
0.9 |
Threshold for calling genotypes
in genotype
input file. The genotype with the maximum probability will be used if
that probability is above the threshold. Otherwise the genotype will
treated as missing. |
| -nind
<int> |
Optional |
Specify the number of
individuals to impute i.e. the impute just the 1st individual use -nind
1 |
|
| -exclude_snps <file> | Optional | Exclude a set of genotyped SNPs
(i.e. SNPs that occur in the file specified by the -g option) with ID
equal to those listed in the file. The IDs can be either the rs ID or
the alternate ID given in the first column of the genotype file. These
SNPs will not be used for imputation and will not occur in the output
files. |
|
| -impute_excluded |
Optional |
This flag modifies the behaviour
of the -exclude_snps
option. For Type 2 SNPs that have been excluded it places imputed
genotypes in the output file. |
|
| -os
<int> -include_snps <file> |
Optional Optional |
1 2 3 |
The SNPs that are included in
the output are controlled by the combination of the -os
and
-include_snps options. The -os option controls which types of SNPs are included in the output. There are three types of SNPs 1 = SNPs that occur ONLY in the haplotypes file 2 = SNPs that occur in BOTH the haplotypes and genotypes file 3 = SNPs that occur ONLY in the genotypes file You can specify more than one type of SNP using the -os option. For example, using -os 1 2 would output SNPs in the haplotypes file. The default setting is to produce output at all snps i.e -os 1 2 3. Using -os 2 is a useful if all you require is an LD-based estimate of the genotypes at SNPs in the genotypes file and can be substantially quicker than the default setting. The -include_snps option specifies a list of SNPs to be included in the output BUT this list only applies to those SNPs that appear only in the haplotype file i.e the SNPs specified by -os 1. The IDs should be the rsIDs given in the legend file that corresponds to the haplotypes file. |
| -pgs |
Optional |
For SNPs that occur in the
genotype file the default is now to return
these genotypes in the output file rather than their predictions (which
was the old default). The -pgs
flag (which stands for predict genotyped snps) can be used to
specify that the predictions should be written to the output file. |
|
| -outdp
<int> |
Optional |
2 |
Specify the number of decimal
places used to report the genotype probabilities. |
| -chrX |
Optional |
Specify this flag if you want to
impute genotypes on the X chromosome. The haplotype files, legend file,
map file and strand file should set appropriately. A sample file must
also be supplied (see -sample
below). |
|
| -Xpar |
Optional |
Controls whether you wish to do
imputation in the pseudoautosomal or non-pseudoautosomal region of the
X chromosome. If the flag is given it specifies that you are working in
the pseudoautosomal region. If the flag is absent it specifies that you
are working in the non-pseudoautosomal region. Only works when used in
conjunction with the -chrX
flag. |
|
| -sample |
Optional |
Sample file (see FILE
FORMAT WEBPAGE for more details) containing a covariate
named 'sex' specifying the sex of all indviduals in the genotype file.
Males should be coded 1 and females coded 2. |
| 0.1.1 | 07-06-2007 | First version
made available |
| 0.2.0 | 29-07-2007 |
|
| 0.2.1 |
22-10-2007 |
Added
LICENCE |
| 0.3.0 |
04-12-2007 |
New
features
|
| 0.3.1 |
17-12-2007 |
|
| 0.3.2 |
18-01-2008 |
|
| 0.4.0 |
26-05-2008 |
New
features
|
| 0.4.1 |
28-05-2008 |
New
feature
|
| 0.4.2 |
16-06-2008 |
New
features
|
| 0.5.0 |
01-08-2008 |
New
features
|