Our approach
(top)
There are many possible ways to merge two reference panels. We are exploring several of these options, but we decided to start with the simple approach depicted in the figure below. The top panel of this figure shows two reference panels and a GWAS cohort; you can think of the rows as individuals and the columns as positions along the genome. Each vertical line represents a genotyped variant in a given panel, and each reference panel includes variants that are not found in the other.
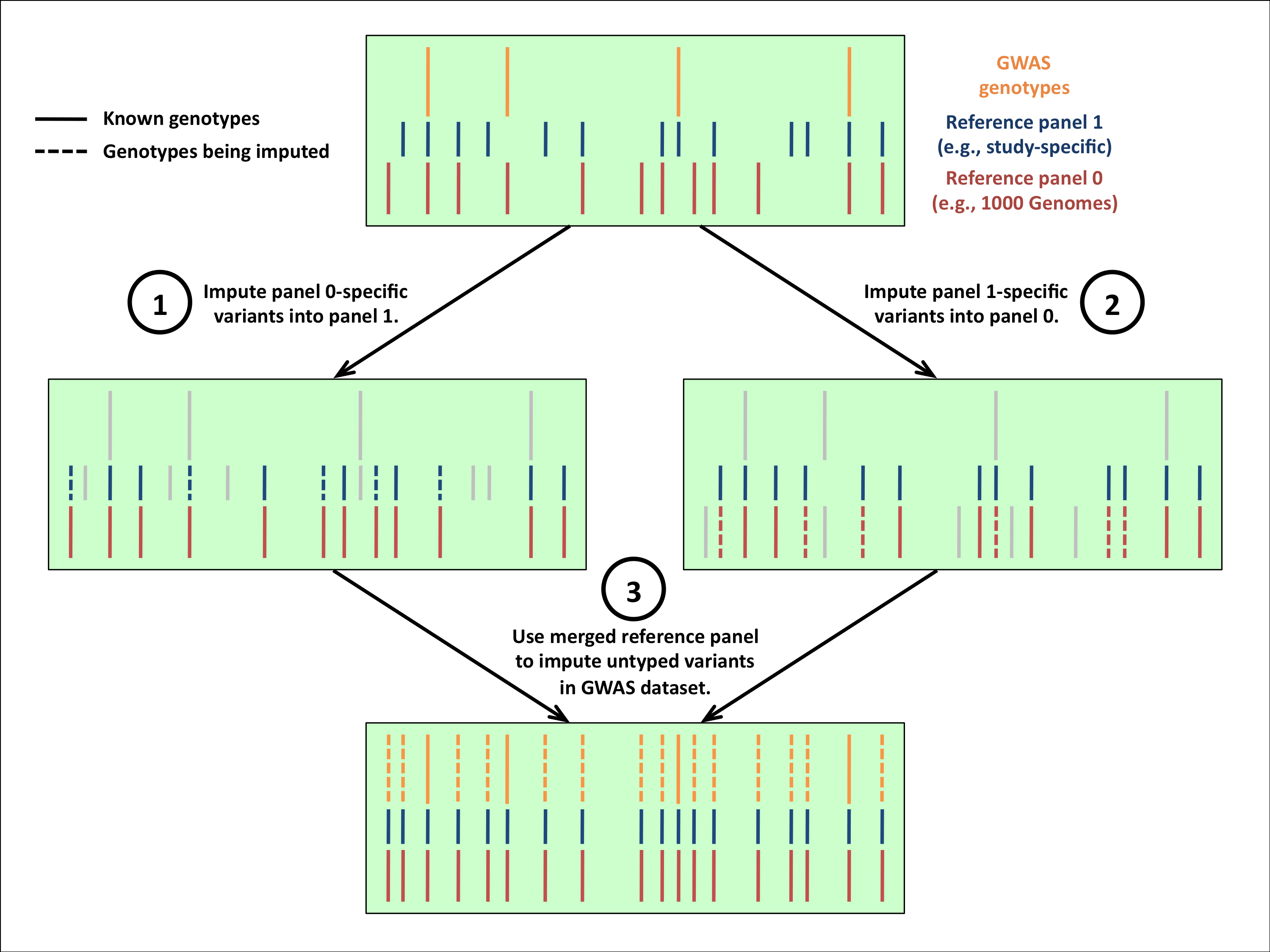
We impute the untyped variants in this figure in three steps:
- Impute the variants that are specific to Panel 0 (red) into Panel 1 (blue). Variants shown in grey do not inform the imputation.
- Impute the variants that are specific to Panel 1 (blue) into Panel 0 (red). Variants shown in grey do not inform the imputation.
- Now that we have imputed the two reference panels up to the union of their variants, treat the imputed haplotypes as known (i.e., take the best-guess haplotypes) and impute the GWAS cohort in the usual way.