How are concordance tables made?
(top)
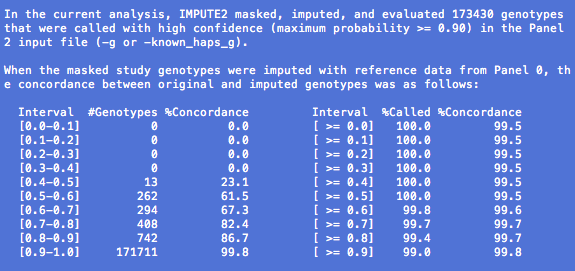
Only variants with input data from a -g or -known_haps_g file are masked and imputed in this analysis. When a -known_haps_g file is provided, all input genotypes are treated as being true. When a -g file is provided, we make hard genotype calls by applying a threshold (default = 0.9) to the maximum value in each input probability triple. For example, a genotype with P(G=0,1,2) = (0.03, 0.95, 0.02) would be called as a '1' (heterozygous), while a genotype with P(G=0,1,2) = (0.1, 0.7, 0.2) would be left uncalled and omitted from the concordance calculations.
The genotype probabilities from imputation are used somewhat differently. In the first three columns of the table, we assign each imputed genotype to a bin (Interval) based on its maximum posterior probability. Then, for each bin we report the number of imputed genotypes that passed the calling threshold in the input data (#Genotypes). We then convert the imputed probabilities to 'best-guess' genotypes: for each posterior probability triple, we select the genotype with the highest value, regardless of magnitude. Finally, we compare the input genotype calls with the best-guess imputed genotypes and report the concordance (%Concordance) within each bin.
In the last three columns of the table, we again bin the imputed genotypes based on their maximum posterior probabilities, but this time the binning is cumulative: the bin at the bottom of the table includes only genotypes that were confidently imputed (max prob >= 0.9), while each bin above includes all genotypes that pass a more lenient certainty threshold. These thresholds are shown in the fourth column (Interval). The fifth column (%Called) shows the percentage of imputed genotypes that pass a given probability threshold, where the denominator is the total number of imputed genotypes for which hard calls are available in the input data. The sixth column (%Concordance) shows the percentage of imputed genotypes in a given bin that match the masked input genotypes.
|