
IMPUTE2
IMPUTE version 2 (also known as IMPUTE2) is a
genotype imputation and haplotype phasing program based on ideas
from
B. N. Howie, P. Donnelly, and J. Marchini (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genetics 5(6): e1000529 [Open Access Article] [Supplementary Material]
IMPUTE2 also includes features that were introduced in other publications, which you can find here.
The figure below shows the most common scenario in which imputation is used: unobserved genotypes (red question marks) in a set of study individuals are imputed (or predicted) using a set of reference haplotypes and genotypes from a SNP chip.
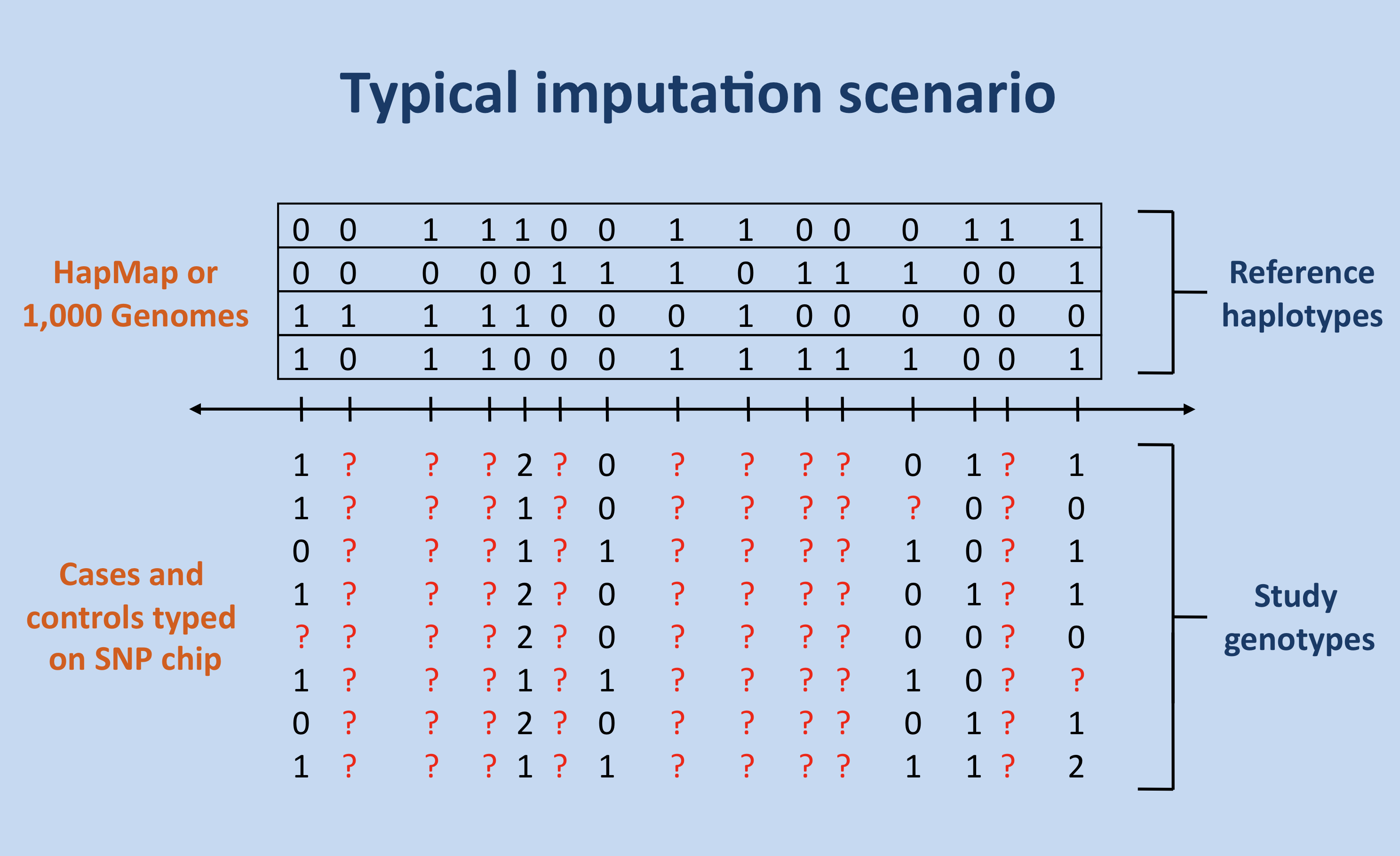
Getting Started
IMPUTE2 is a computer program for phasing observed genotypes and imputing missing genotypes. Most people use just a couple of the program's basic functions, but we have also built up a collection of specialized and powerful options. If you are new to IMPUTE2, or indeed to phasing and imputation in general, we suggest that you start by learning the basics.
You should begin by downloading the program from here. You will need to choose the link that matches your computing platform and then follow the instructions for opening the download package.
Once you have done this, you will be ready to try some example
analyses on the test data that are provided with the download.
The section on
When you have learned the basic functionality of the program, you can use several features of this website to prepare your own analysis:
- Learn about best practices for imputation.
- Download reference data that you can use to impute genotypes in your study.
- Look through a complete list of program options.
What's New?
New release (23 December 2014)
We have just released IMPUTE v2.3.2. This version is a
very minor update to add additional columns that report the two
alleles at each imputed variant to the info files.
New release (16 June 2014)
We have just released IMPUTE v2.3.1. This version fixes
a bug in panel-merging
functionality that caused variants seen in one of two
reference panels to be imputed with a fixed allele (non-ref
allele in Panel 0, ref allele in Panel 1). If you have used
these options, then we would recommend re-running your
imputation.
In addition, we have released a new version of the 1000 Genomes Phase 1 haplotypes. These are an updated version of the haplotypes released on 9 Dec 2013. It seems we had not completely resolved the strand flip issue with the previous release (see below), and a further 199 SNPs needed to be corrected in this new release.
The new haplotypes are available here.
New release (9 Dec 2013)
We have released a new version of the 1000 Genomes Phase 1
haplotypes. These are an updated
version of the haplotypes released on 16 Sept 2013. There was a small problem
with the strand of the Illumina OMNI data we used as the
scaffold. 730 SNPs across the genome were not aligned to the +
strand of the human genome reference. This does not affect the
phasing of the haplotypes, but does affect downstream
imputation, especially if these SNPs were genotyped directly
in the study being imputed. The new haplotypes were not
re-phased. We just switched the strand of the 730 affected
SNPs.
The new haplotypes are available here.
New release (16 Sept 2013)
We have released a new version of the 1000 Genomes Phase 1
haplotypes. The
haplotypes were phased using a new version of SHAPEIT2
that can handle genotype likelihoods and genotypes available
from microarrays on the same samples. Using a set of validation
genotypes at SNP and biallelic indels we have been able to
show that these haplotypes have lower genotype discordance and
improved imputation performance into downstream GWAS samples,
especially at low frequency variants.
The new haplotypes
are available here.
New software release (04 Jan 2013)
We have just released IMPUTE v2.3.0, which includes a number of new features and minor bug fixes. One valuable new function is a simple and robust approach for merging reference panels; for example, it is easy to combine 1,000 Genomes haplotypes with population-specific sequence data to capture the strength of both reference sets. We have also written detailed documentation for the concordance tables printed at the end of most IMPUTE2 runs.
Paper on "pre-phasing" study genotypes for faster imputation
We recently published an article called "Fast and accurate genotype imputation in genome-wide association studies through pre-phasing" in Nature Genetics. This paper describes a strategy ("pre-phasing") for efficient genotype imputation with large reference panels. By reducing the computational burden of imputation, pre-phasing makes imputation-based studies feasible for groups with limited computing power, and it also makes it easier to re-impute existing GWAS datasets as more informative reference panels become available. You can learn more about pre-phasing with IMPUTE2 here.
Latest 1,000 Genomes Phase I reference panel
In March 2012, the 1,000 Genomes Project released a powerful reference panel known as "Phase I version 3". In August 2012, we modified this panel by excluding variants with only one copy of the minor allele (singletons) across all 1,092 individuals. Singleton variants are difficult to impute, yet they make up ~20% of all variants in the reference panel; removing them makes imputation faster without hurting the power for association mapping. You can download either the orginal reference panel or the modified version (which is labeled "macGT1" for "minor allele count greater than one") here.
Paper on imputation strategies for ancestrally diverse reference panels
We published an article called "Genotype
imputation with thousands of genomes" in the open-access
journal G3: Genes,
Genomes, Genetics. This paper describes our strategy
for achieving high accuracy with ancestrally diverse reference
panels, especially at low-frequency variants and in admixed
study cohorts: we supply a cosmopolitan set of reference
haplotypes to IMPUTE2, which can automatically find the
most useful ones for each study individual with the help of the
tuning parameter
Pre-phasing with SHAPEIT
IMPUTE2's pre-phasing approach now works with phased haplotypes from SHAPEIT, a highly accurate phasing algorithm that can handle mixtures of unrelateds, duos, and trios. Details are available here. We highly recommend using SHAPEIT to infer the haplotypes underlying your study genotypes, then passing these to IMPUTE2 for imputation as shown in the second step of this example.
Download IMPUTE2
IMPUTE2 is freely available for academic use. To see rules for non-academic use, please read the LICENCE file, which is included with each software download.
Pre-compiled IMPUTE2 binaries and example files can be downloaded from the links below. For Linux machines, the dynamic binaries are smaller but may not work on some machines due to gcc library compatibility issues; if the dynamic version doesn't work for you, please try the static version. If you have any problems getting the program to work on your machine or would like to request an executable for a platform not shown here, please send a message to our mail list.
The latest software release is v2.3.1. We support only the most recent version.
| Platform | File |
|---|---|
| Linux (x86_64) Static Executable | impute_v2.3.2_x86_64_static.tgz |
| Linux (x86_64) Dynamic Executable | impute_v2.3.2_x86_64_dynamic.tgz |
| Mac OSX Intel | impute_v2.3.2_MacOSX_Intel.tgz |
| Windows MS-DOS (Intel) | impute_v2.3.1_Windows.tgz (coming soon) |
| Solaris 5.10 | impute_v2.3.2_Solaris5.10.tar.gz (coming soon) |
To unpack the files on a Linux computer, use a command like this:
tar -zxvf impute_v2.X.Y_i386.tgz
(Other file decompression programs are available for non-Linux
computers.) This will create a directory of the same name as the
downloaded file, minus the '.tgz' suffix. Inside this directory
you will find an executable called impute2,
a LICENCE file, and an Example/ directory that contains example
data files. We show how to perform various kinds of analyses
with the example files here.
Download Reference Data
IMPUTE2 can use publicly available reference datasets, such as haplotypes from major sequencing projects, as well as customized reference panels, such as SNP genotypes from a fine-mapping study. If you would like to download a public dataset, just click the relevant link below, which will take you to a page with background information and download options for that dataset.
| |
|
|
|
|---|---|---|---|
| 1000
Genomes Phase 3 |
b37 |
October 2014 |
|
| 1000 Genomes Phase I integrated haplotypes (produced using SHAPEIT2) | b37 |
June 2014 |
|
| 1000 Genomes Phase I integrated haplotypes (produced using SHAPEIT2) | b37 | Dec 2013 |
|
| 1000
Genomes Phase I integrated haplotypes (produced using
SHAPEIT2) |
b37 |
Sep 2013 |
|
| 1000 Genomes Phase I integrated variant set | b37 | Mar 2012 | Includes chrX; updated 24 Aug 2012 |
| 1000 Genomes Phase I (interim) | b37 | Jun 2011 | Includes chrX; updated 19 Apr 2012 |
| 1000 Genomes (2010 interim) | b37 | Dec 2010 | |
| 1000 Genomes Pilot + HapMap 3 | b36 | Jun 2010 / Feb 2009 | |
| 1000 Genomes Pilot | b36 | Jun 2010 | |
| HapMap 3 (release #2) | b36 | Feb 2009 | Includes chrX |
| HapMap 2 (release #24) | b36 | Oct 2008 | |
| HapMap 2 (release #22) | b36 | Jan 2008 | |
| HapMap 2 (release #21) | b35 | Jul 2006 | |
Using Multi-Population Reference Panels
Overview
Human genetic variation resources, like those produced by HapMap 3 and the 1,000 Genomes Project, capture a broad cross-section of human genetic diversity: detailed variation data have now been collected from a variety of sampling locations in Africa, Asia, Europe, and the Americas. Large sequencing projects are actively expanding these datasets to include additional populations and deeper sampling within populations. These public databases provide powerful reference panels for genotype imputation studies.
In this context, one important question is how to choose a reference panel that will produce high imputation accuracy in a population of interest. The answer is seldom obvious because human populations have experienced complex demographic histories with many migration and mixture events. Consequently, it can be hard to decide which reference haplotypes should be used in a particular study.
We have proposed a simple and universal solution to this problem: we provide all available reference haplotypes to IMPUTE2, then let the software choose a "custom" reference panel for each individual to be imputed. There are several advantages to this approach:
- Investigators do not need to waste time
deciding which haplotypes to include in the reference panel.
Good results can be obtained in any study population by tuning
a single software parameter (
-k_hap ) with a simple rule of thumb; see below for more details. - This strategy works in a variety of human populations. Our group and others have used this approach to successfully impute populations ranging from homogeneous isolates to recent and complex admixtures.
- IMPUTE2 is often more accurate
with an ancestrally inclusive reference panel than with a
smaller panel chosen by intuition. This is because
individuals from "diverged" populations may still share
genomic segments of recent common ancestry, and IMPUTE2
can use this haplotype sharing to improve accuracy. At the
same time, the software can ignore haplotypes that are not
helpful.
The benefits of using inclusive reference panels are greatest at low-frequency variants (MAF < 5%), since these variants may be poorly represented in a reference panel from the population of interest (due to sampling effects) but well-represented in panel from a different population (e.g., due to genetic drift). - IMPUTE2 can efficiently process large reference panels. You might worry that using all available reference haplotypes would greatly increase the computational burden of imputation, but IMPUTE2 uses an approximation that limits the cost of adding reference haplotypes while maintaining (or improving) accuracy.
Practical suggestions
There are a few program settings that you should be aware of when using IMPUTE2 with an ancestrally diverse reference panel:
-
-k_hap �This parameter determines how many of the reference haplotypes will be used in the "custom" reference panel for each study individual. The default value is 500, which is a good starting point for modern reference datasets.
As a rule of thumb, you should set -k_hap to the number of reference haplotypes that you expect to be useful for your study population. For example, suppose you were imputing a Spanish dataset from a reference panel containing 400 Western European haplotypes and 400 African American haplotypes. In this case, you could achieve high accuracy by leaving-k_hap at the default value of 500 since, in any part of the genome, the expected number of reference haplotypes with European ancestry is roughly400 + 0.2 * (400) = 480 . (This calculation assumes that, on average, African American haplotypes have 20% European ancestry.)
Imputation accuracy is not very sensitive to-k_hap , which is why this rule of thumb usually provides good results without requiring detailed parameter tuning. If you want advice on the best value for your dataset, please send a message to our mail list. -
-Ne �This parameter controls the effective population size in the population-genetic model used by IMPUTE2. Different human populations have different effective sizes (as estimated from genetic diversity levels), so it is not obvious how to choose a single-Ne value when using a multi-population reference panel.
Fortunately, we have found that IMPUTE2 achieves high accuracy across a wide range of-Ne values, with slightly higher accuracy at large values. We therefore recommend a universal-Ne value of 20000, regardless of the study population being imputed or the composition of the reference panel. This will become the default value in our next software release (v2.1.3), but for now you should set it manually. -
-int �This command-line option specifies the boundaries of the region to be imputed on the current chromosome, using two numbers. For example,"-int 1 5e6" tells IMPUTE2 to analyze physical positions 1-5,000,000.
The imputation interval should not be too large because this weakens IMPUTE2's approximation for choosing custom reference panels, which is based on an assumption of limited recombination in the region being analyzed. In theory, it might be desirable to tailor the interval size to the population being imputed�e.g., to use shorter intervals in African populations�but in practice, we have found that the exact size of the interval has little effect on imputation accuracy as long as the interval is relatively small (say, < 10 Mb). We therefore recommend that the size of the analysis interval be chosen for computational convenience, without regard to the ancestry of the study or reference datasets.
How does it work?
As explained above, we believe that the best way to use IMPUTE2 with modern reference panels is to provide all available haplotypes to the program and let it choose which ones to use. Here, we explain how this approach works.
IMPUTE2 does not use population labels or other genome-wide measures of relatedness between individuals, either for the reference haplotypes or the individuals being imputed. Instead, it looks for reference haplotypes that share high sequence identity with the haplotypes of a particular study individual. These haplotypes constitute a "custom" reference panel that can be used to impute missing genotypes in the individual of interest.
This process is largely insensitive to the ancestral composition of the reference panel: as long as the panel contains haplotypes that share segments of recent common ancestry with individuals in a study, IMPUTE2 can find the shared segments and use them to impute missing alleles. Consequently, the reference panel does not need to be restricted to haplotypes that "match" the ancestry of the study individuals�it can also include other kinds of haplotypes:
- Recently admixed haplotypes�If two or more distinct populations have mixed within the past few hundred years, the resulting admixed population may contain some haplotype segments that are closely related to a population of interest and other segments that are highly diverged. IMPUTE2 can identify the useful segments while ignoring the diverged segments, thereby achieving accurate imputation.
- Moderately diverged haplotypes�Even if a set of reference haplotypes comes from a different population than the one you want to impute, it may still provide segments of recent ancestry that can help the imputation. The prevalence of such segments is a complicated function of reference panel size and population history, but in our experience there is often a surprising amount of ancestry sharing between genetically distinct populations.
- Highly diverged haplotypes�Reference haplotypes that are highly diverged from your study population are unlikely to be useful for imputation, but such haplotypes are easily identified and ignored by IMPUTE2. In other words, highly diverged reference haplotypes neither help nor hurt imputation accuracy. This is important because the distinction between "moderately" and "highly" diverged populations is not always clear; since it does not hurt to include unhelpful reference haplotypes, we can err on the side of including too many in order to capture more of the moderately diverged ones that improve imputation accuracy.
Expert users will note that the model underlying IMPUTE2 is formally designed to represent genetic variation in a single population. This might imply that the method would have trouble using reference panels that include populations with different linkage disequilibrium patterns, nucleotide diversity levels, and allele frequency spectra. However, we have found that the IMPUTE2 is extremely adaptable: it can find segments of shared ancestry in multi-population reference panels despite its simple model of human populations, and it is largely robust to changes in its model parameters. Imputation accuracy might theoretically be improved by more detailed modeling of population relationships (for example, the population labels that IMPUTE2 ignores might sometimes be informative), but we believe that our approach captures most of the potential accuracy in an efficient way.
Published results
We published our work supporting these ideas in an article called "Genotype imputation with thousands of genomes" in the open-access journal G3: Genes, Genomes, Genetics. Please cite this paper and the original IMPUTE2 paper when using IMPUTE2 with multi-population reference panels like those from the 1,000 Genomes Project.
Examples
This section provides some example commands that illustrate typical applications of IMPUTE2. All of the data files used in these commands are included in the Example/ directory that comes with the software download. You should run the commands from the main download directory (i.e., the one that contains the impute2 executable). Detailed explanations are provided at each link below.
| Run type | Description |
|---|---|
| Imputation with one phased reference panel | Basic scenario in which most people will use IMPUTE2. |
| Imputation
with one phased reference panel (pre-phasing) |
As above, but with pre-phasing functionality to speed up the analysis. |
| Imputation
with one phased reference panel (chromosome X) |
Basic imputation scenario applied to human chromosome X, which requires special program options. |
| Imputation
with one phased reference panel (plus variant filtering) |
Basic imputation scenario with flexible filtering of reference panel variants. |
| Imputation with one unphased reference panel | Basic imputation scenario adapted to unphased reference genotypes. |
| Imputation with two phased reference panels | Extended functionality for imputing from multiple reference panels defined on different sets of variants. |
| Imputation
with two phased reference panels (merge reference panels) |
Merge reference panels defined on different sets of variants and use combined panel for imputation. |
| Imputation with one phased and one unphased reference panel | Specialized method for combining reference panels of different types. |
| Imputation with one phased and one unphased reference panel, with additional options | As above, but illustrating a variety of options that can be used to customize the behavior of IMPUTE2. |
| Phasing | Methodology for inferring haplotypes from unphased genotypes. |
| Phasing with a reference panel | Phasing analysis aided by reference haplotypes. |
How to use example commands
All of the data files in the example commands below are
included in the Example/
directory that comes with the IMPUTE2 software download.
You should run the command from the main download directory,
which is the one that contains the impute2
executable. For example, if you just downloaded a software
package named impute_v2.X.Y_i386.tgz
and unpacked it according to the directions here,
you can reach the appropriate directory by typing "
Once you have found the right directory, you should be able to run the example command by entering it into a Unix-style terminal window. Depending on the settings of your computer, this may be as simple as highlighting the command text in your web browser, using the browser's Copy command, and then using the Paste command in your terminal window. (You may then need to hit Enter to start the run.)
Note that most lines in the example command end with the '\' character. This is not actually part of the command; it is just a shorthand notation that means "keep reading the next line as part of a single command." We use this notation to split the command over multiple lines so it is easier to read. This is a valid way to enter commands in a Unix-style terminal window, but it would be equivalent to put all of the arguments on a single line, separated by spaces.
You do not have to run IMPUTE2 exactly as in the example. Some of the arguments shown here are optional, and there are many other options that could be added to modify the behavior of the program. For a full list of available options, see here.
Most of the examples below include the string "
Imputation with one phased reference panel
This is the most common genotype imputation scenario: we want to impute untyped SNPs in a study dataset from a panel of reference haplotypes.
The following command shows how to run this kind of analysis with IMPUTE2, using the example data that come with the program download:
./impute2 \
Comments
- Here we have used the
-strand_g option to provide a strand file to the program. This file tells IMPUTE2 how to align the allele coding between the study genotypes (-g file) and the reference haplotypes (-h and-l files). You must always align the allele codings across your input datasets, either before running IMPUTE2 or during a run with the options described here. - This command invokes the standard MCMC algorithm used by IMPUTE2,
which usually provides accurate results in a reasonable amount
of time. Another way to run this kind of analysis is to use
our
pre-phasing approach , which decreases the running time by orders of magnitude at the cost of a small drop in imputation accuracy. To see how to run this example with pre-phasing, click here.
Imputation with one phased reference panel (pre-phasing)
This is the most common genotype imputation scenario: we want
to use a panel of reference haplotypes to impute SNPs that were
not typed in a study. Here, we show how to perform this task via
The following commands show how to run this kind of analysis with IMPUTE2, using the example data that come with the program download:
Step 1: Pre-phasing
./impute2 \
Step 2: Imputation into pre-phased haplotypes
./impute2 \
-phase
Comments
- Pre-phasing is a useful technique for speeding up an imputation run, but it is even more useful if you want to impute a single study dataset from different reference panels (e.g., successive updates to the reference haplotypes released by the 1,000 Genomes Project). In that situation, you can perform the pre-phasing step just once and save the estimated haplotypes; you can then use the same study haplotypes to perform the imputation step with each new reference panel.
- If you are using IMPUTE2 for both the pre-phasing
and subsequent imputation, it is important to use the same
values of the
-int parameter in both steps. - The
-prephase_g flag activates a couple of features that are necessary for pre-phasing. First, it tells the program to estimate and print phased haplotypes at SNPs included in the-g file; the haplotypes will be written to a file named"[-o]_haps" , where[-o] is the name supplied for the main output file. These haplotypes will include SNPs in the buffer regions that flank the main region specified via-int . Extending the haplotypes into the buffer regions helps prevent edge effects in downstream imputation runs. - It is possible to include a reference panel in the
pre-phasing step, and this may improve the phasing quality.
See
here for an example of this kind of analysis (note that the linked example is missing the-prephase_g flag). To expedite the pre-phasing in this scenario, the program will not impute reference-only variants when-prephase_g is active, although you can override this behavior with the-os option. - You can use the
-strand_g option in either the pre-phasing or downstream imputation step, but you should not use it in both. Strand alignment is not usually necessary when you just want to phase a dataset, but it is important when that dataset will be combined with a reference panel in a downstream analysis, as in this case. - Note that the file supplied to the
-known_haps_g argument in the imputation step is the estimated haplotypes file from the pre-phasing step ("[-o]_haps" ). Also note that the-use_prephased_g flag must be provided when imputing into pre-phased haplotypes. - The -phase option in Step 2 above produces a file containing the haplotypes at the imputed and genotyped sites. In this example, the file would be called example.chr22.one.phased.impute2_haps
- Pre-phasing based imputation on chromosome X is also
possible. The only things you need to do differently are to
make sure that you supply a sample file for your data using
the
-sample_g option (so IMPUTE2 knows which individuals are male and which are female), and use the-chrX flag, and ensure that your male haploid genotypes are encoded according to the described file format. If you use SHAPEIT2 for the phasing step then this software also has options to phase chromosome X. Below is an example of how to carry out chromosome X pre-phasing based imputation
Step 1: Chromosome X Pre-phasing
./impute2 \
-prephase_g \
-chrX \
-m
./Example/chrX/example.chrX.map \
-sample_g
./Example/chrX/example.chrX.study.sample \
Step 2: Imputation into pre-phased chromosome X
haplotypes
./impute2 \
-use_prephased_g \
-chrX \
-phase
Imputation with one phased reference panel (chromosome X)
This example provides a twist on the common scenario of imputing untyped SNPs in a study dataset from a panel of reference haplotypes. Here, we want to perform the analysis on chromosome X, which requires special treatment due to the hemizygosity of males. (This example and the files in our download packages focus on the non-pseudoautosomal part of chromosome X.)
The following command shows how to run this kind of analysis with IMPUTE2, using the example data that come with the program download:
./impute2 \
Comments
- The
-chrX flag is essential because it tells IMPUTE2 to expect the special file formatting conventions used for chromosome X data. - Whenever you analyze data on chromosome X, you must also
provide a
-sample_g file so that the program knows which individuals are males and which are females. You can learn about the specific requirements of this file here. - There is no need to use a different
-Ne value on chromosome X than you would on the autosomes; the-chrX flag tells IMPUTE2 to automatically reduce the value by 25%, which changes the parameters of the haplotype copying model. - Like the input files, the IMPUTE2 output files from chromosome X analyses should be interpreted according to these conventions.
File formats for chromosome X
Among human chromosomes, chromosome X is unique in that it is
dizygous (two copies) in females but hemizygous (one copy) in
males. To deal with chromosome X data, IMPUTE2 requires
that you use the
- Genotypes file (
-g ): As in a standard-g file, each study individual should have three columns (genotype probabilities) per SNP. For females, these have the standard interpretation that columns 1, 2, and 3 represent P(G=0), P(G=1), and P(G=2), respectively, where G=1 is the heterozygous state. Males have only two possible genotypes on chromosome X, and we encode these in columns 1 and 3; column 2, which corresponds to P(G=1), should always be zero in this setting, and non-zero values in this column will automatically be truncated to zero for males when the-chrX flag is active. - Sample file (
-sample_g ): In order for the input genotype convention explained above to work, IMPUTE2 needs to know which study individuals are males and which are females. This is accomplished by adding an extra column named'sex' to the-sample_g file, which is required when using the-chrX flag. This column should be coded as type'D' (discrete covariate), where males are indicated by'1' s and females are indicated by'2' s. Here is an example snippet where the first individual is female and the second and third individuals are male:ID_1 ID_2 missing sex
0 0 0 D
INDIV1 INDIV1 0.0 2
INDIV2 INDIV2 0.0 1
INDIV3 INDIV3 0.0 1
- Reference haplotypes file (
-h ): It does not usually matter which reference individuals are male or female when their genotypes have already been phased. However, it may sometimes be convenient to create a-h file with two columns per individual, so IMPUTE2 allows the presence of dummy columns made of'-' characters to represent the non-existent second haplotypes of males on chromosome X. For example, here is a small haplotypes file with 5 SNPs (one per row) typed in a female (columns 1-2) and two males (columns 3-4 and 5-6):0 1 1 - 0 -
The dummy columns are optional�the following would be an equally valid format for the same file:
0 0 1 - 1 -
1 0 0 - 1 -
1 1 0 - 1 -
0 0 1 - 0 -
0 1 1 0
0 0 1 1
1 0 0 1
1 1 0 1
0 0 1 0
- Output files (
-o ): The main output file will follow the same convention as the genotypes file described above: each individual has three entries per SNP, but the middle entry is set to zero for males. When IMPUTE2 produces haplotype output files for chromosome X, both males and females will have two columns per individual, although the second column for each male will be filled with dummy values of'-' .
Imputation with one phased reference panel (plus variant filtering)
This example provides a twist on the common scenario of imputing untyped SNPs in a study dataset from a panel of reference haplotypes. Here, we want to perform the analysis after flexibly removing a subset of sites from the reference panel.
The following command shows how to run this kind of analysis with IMPUTE2, using the example data that come with the program download:
./impute2 \
Comments
- The main novelty here is the use of the
-filt_rules_l option. This option works by defining "filtering rules" that combine annotation categories (here,eur.maf andafr.maf and TYPE) with comparison operators (< and<= and == ) and values (0.01 and0.05 and LOWCOV). Each annotation string is present on the first line of the-l file and is followed by a column of numeric or character values (one for each site in the reference panel) that determine whether a given site should be filtered from the reference set. In this example, the filtering rules tell IMPUTE2 to ignore reference variants with minor allele frequency less than 1% in a European panel OR less than 5% in an African panel OR sites that are annotated as LOWCOV (Filtering rules are always applied in 'OR' fashion.) - You can make your own filtering rules by adding numeric or
character annotation columns to a reference legend (
-l ) file, or you can use the annotations that we provide in some of our reference panel download packages. For example, we have included continent-level minor allele frequencies in the legend files for the 1,000 Genomes Phase 1 integrated variant reference panel. - USAGE GUIDELINES FOR FILTERING
RULES: Our main motivation in creating the
-filt_rules_l option was to provide a fast and easy way of reducing the computational burden of large, sequence-based reference panels. A principled way to do this is to remove the reference SNPs that are expected to provide the least power in an imputation-based association analysis. We suggest that the rarest SNPs in a dataset fall into this category, both because there is generally less power to detect these under many study designs and because such SNPs are often harder to impute, which further diminishes the real power for detection. So, one simple approach is to use a minor allele frequency filtering rule (e.g.,'eur.maf<0.01' ) for MAF annotations from a population like the one being studied.
Imputation with one unphased reference panel
It is not necessary for the reference panel to be phased: IMPUTE2
can do the phasing internally while accounting for the phase
uncertainty. To use an unphased reference panel, simply replace
the
The following command shows how to run this kind of analysis with IMPUTE2, using the example data that come with the program download:
./impute2 \
Comments
- As with any imputation analysis, it is
important that all of your input files be aligned to the
same allele coding at shared SNPs. In this example,
we assume that both the
-g_ref and-g files include SNPs that are not aligned to the '+' strand of the human genome reference sequence, so we use the-strand_g_ref and-strand_g options to bring them into alignment. - This procedure is not recommended for unphased reference panels that have high SNP density, such as those that result from resequencing studies of population samples. In that situation, there may be statistical convergence issues that could decrease the imputation quality. If you need advice on how to use that kind of reference dataset, please send a message to our mail list.
Imputation with two phased reference panels
It is sometimes helpful to use multiple reference panels to impute genotypes in a single study. For example, we previously recommended combining reference haplotypes from the 1,000 Genomes Pilot Project and HapMap 3: the first set provided extensive coverage of polymorphisms in the genome, while the second set provided greater sample size at a subset of SNPs. We no longer recommend that you use this hybrid reference panel because the 1,000 Genomes Project has generated even richer reference sets (which you can download here), but some investigators may have additional reference data that could be used in this way.
The following command shows how to run this kind of analysis with IMPUTE2, using the example data that come with the program download:
./impute2 \
Comments
- This is a somewhat complicated scenario, and some restrictions are necessary to make sure the statistical machinery will produce good results. Ideally, one reference panel should contain a subset of the SNPs typed in the other reference panel, and the study dataset should contain a subset of the SNPs typed in both reference panels. If your dataset deviates substantially from these conditions, you may obtain sub-optimal imputation accuracy. Please send a message to our mail list if you want advice on whether this scheme will work with your data.
- Assuming the conditions described above are nearly
satisfied, the reference panel with a larger number of SNPs
should always come first on the command line. In this example,
there are more SNPs in the 1,000 Genomes ("1kG") panel than in
the HapMap 3 ("hm3") panel, so the 1,000 Genomes files
are listed first after the
-h and-l arguments. - Here we have used the
-strand_g option to provide a strand file to the program. This file tells IMPUTE2 how to align the allele coding between the study genotypes (-g file) and the reference haplotypes (-h and-l files; assumed to be aligned to the '+' strand of the human genome reference sequence). You must always align the allele codings across your input datasets, either before running IMPUTE2 or during a run with the options described here.
Imputation with two phased reference panels (merge reference panels)
Many investigators have access to multiple reference panels that could inform their imputation analyses. For example, they might want to supplement the 1,000 Genomes haplotypes (which can be downloaded here) with dedicated sequencing data from a study population.
If you have two panels that have been phased and put into IMPUTE2's reference format (legend/haplotype file pairs), you can ask the program to merge them internally and impute your study genotypes by entering the following command, which uses example data that come with the program download:
./impute2 \
Comments
- For details on how the reference panel merging works, please read the documentation.
- This approach also works with pre-phased study haplotypes.
To use pre-phased study data in this example, you would
replace the
-g file with a-known_haps_g file and add the-use_prephased_g flag to your IMPUTE2 command. - If you want to print the merged, phased panel in IMPUTE2
reference format (one
-l file and one-h file), you should add the-merge_ref_panels_output_ref flag. - If you want to print the merged, unphased panel in IMPUTE2
genotype format (one
-g file), you should add the-merge_ref_panels_output_gen flag. - If you simply want to merge two reference panels without
imputing missing genotypes in a study dataset, you should add
the
-merge_ref_panels_output_ref or-merge_ref_panels_output_gen flag and omit the study genotypes (-g or-known_haps_g file) from your IMPUTE2 command.
Imputation with one phased and one unphased reference panel
Sometimes it is useful to combine a phased reference panel
with an unphased reference panel when imputing genotypes in a
study. For example,
The following command shows how to run this kind of analysis with IMPUTE2, using the example data that come with the program download:
./impute2 \
Comments
- This is a somewhat complicated scenario, and some
restrictions are necessary to make sure the statistical
machinery will produce good results. Ideally, the study data (
-g file) should contain a subset of the SNPs in the unphased reference panel (-g_ref file), which should in turn contain a subset of the SNPs in the phased reference panel (-h and-l files). If your dataset deviates substantially from these conditions, you may obtain sub-optimal imputation accuracy. Please send a message to our mail list if you want advice on whether this scheme will work with your dataset. - Here we have used the
-strand_g and-strand_g_ref options to provide strand files to the program. These files tell IMPUTE2 how to align the allele coding of the study genotypes (-g file) and the unphased reference genotypes (-g_ref file) with the coding of the phased reference haplotypes (-h and-l files; assumed to be aligned to the '+' strand of the human genome reference sequence). You must always align the allele codings across your input datasets, either before running IMPUTE2 or during a run with the options described here. - Additional options must be invoked if you want to include
the
-g_ref panel in your association tests (e.g., as part of your control set). This process requires a fair amount of imputation expertise, and we prefer to advise people about it on an individual basis. If you are interested in using this approach, please send a message to our mail list.
Imputation with one phased and one unphased reference panel, with additional options
Here we perform the same basic analysis as in this example, but we use a number of additional options to modify the behavior of IMPUTE2.
The following command shows how to run this kind of analysis with IMPUTE2, using the example data that come with the program download:
./impute2 \
Comments
- These comments will focus on the specialized options used in the example above; for comments on this general imputation scenario, see here.
- The
-exclude_snps_g_ref option specifies a few SNPs to remove from the-g_ref file, using different types of SNP IDs. These might be SNPs that failed QC testing, for example. - The
-align_by_maf_g option tells the program to use minor allele frequencies to align the allele coding of A/T and C/G SNPs between the-g file and the-l file. However, the-strand_g option takes precedence over-align_by_maf_g , and in this case all of the genotyped SNPs have explicit alignments in the strand file, so the-align_by_maf_g flag has no effect. - This run includes both a
-sample_g file and an-exclude_samples_g file. The sample file tells IMPUTE2 which samples in the-g file are which, and the exclusions file tells it the IDs of samples that should be removed from the analysis. These might be individuals who showed systematic data quality problems on a genome-wide SNP chip, for example. - Here we have increased
-k from its default value of 80 to 100. This will increase the imputation accuracy, but it will also increase IMPUTE2's running time. In this example we have tried to offset the increased running time by decreasing the-burnin value from 10 (default) to 5 and the-iter value from 30 (default) to 20. - The
-pgs flag tells the program to "predict genotyped SNPs"; that is, to replace the original study genotypes with LD-based imputed genotypes in the output file. - The
-no_sample_qc_info flag suppresses the output file that shows quality control metrics for each individual in the-g file. - The
-o_gz flag specifies that the main output file should be compressed by the gzip algorithm; this is useful if you are running jobs that produce large output files.
Phasing
Although IMPUTE2 was originally designed to impute
missing genotypes, it can also be used for a classical phasing
analysis in which we want to infer the haplotypes underlying a
set of observed genotypes. This functionality is activated via
the
The following command shows how to run this kind of analysis with IMPUTE2, using the example data that come with the program download:
./impute2 \
Comments
- The
-o file is always reserved for imputation output, so the phased haplotypes in this example get printed to a file named./Example/example.chr22.phasing.impute2_haps , where the_haps suffix is added automatically. The format of this output file is explained here. - No strand alignment is needed in this example since we are
using only one data panel. However, it may
be important to align the strand at this stage if you intend
to use the phased haplotypes for downstream imputation, i.e.
in a
pre-phasing analysis. - In our experience this phasing procedure works well for SNP chip data, but it may have statistical convergence issues in datasets with high marker density, such as those that result from resequencing studies of population samples. If you would like to phase that kind of dataset, please send a message to our mail list for suggestions about how to improve the quality of inference.
We have not yet posted instructions for how to reattach phased haplotypes across successive chunks along a chromosome. If you want to try this approach to phasing a whole chromosome, please send a message to our mail list.
Phasing with a reference panel
Although IMPUTE2 was originally designed to impute
missing genotypes, it can also be used for a classical phasing
analysis in which we want to infer the haplotypes underlying a
set of observed genotypes. This functionality is activated via
the
Here, we extend a basic phasing analysis to incorporate a phased reference panel. Population-based phasing methods work by pooling linkage disequilibrium information across individuals, so adding a panel of high-quality haplotypes can improve phasing accuracy.
The following command shows how to run this kind of analysis with IMPUTE2, using the example data that come with the program download:
./impute2 \
Comments
- The
-o file is always reserved for imputation output, so the phased haplotypes in this example get printed to a file named./Example/example.chr22.phasing.impute2_haps , where the_haps suffix is added automatically. The format of this output file is explained here. - The reference panel in this example includes SNPs that are
not present in the
-g file. IMPUTE2 can simultaneously impute the untyped SNPs and phase the typed SNPs in that file, but it will not phase the untyped SNPs; the main output file (./Example/example.chr22.phasing.impute2 ) will include estimated genotypes for all study + reference SNPs, but the phased haplotype output file (./Example/example.chr22.phasing.impute2_haps ) will include only the SNPs from the-g file. We decided not to have the program produce haplotypes at reference-panel-only SNPs because the computation needed to provide good estimates is much greater than that needed to phase just the input genotypes or to impute the untyped SNPs without phasing them. If you really want to try phasing the untyped SNPs as well, please send a message to our mail list. - If you don't care about imputing the reference-panel-only
SNPs into your study data (i.e., you just want to phase the
original genotypes), you can substantially speed up the
inference by adding "
-os 2 " to the command line. This tells the program to "output SNPs of type 2", which are ones with input data in both the reference and study panels. By implicitly telling the program not to output other kinds of SNPs (e.g., those typed only in the reference panel), you allow it to avoid wasting calculations that won't contribute to the final output. - Here we have used the
-strand_g option to provide a strand file to the program. This file tells IMPUTE2 how to align the allele coding between the study genotypes (-g file) and the reference haplotypes (-h and-l files). You must always align the allele codings across your input datasets, either before running IMPUTE2 or during a run with the options described here. - In our experience this phasing procedure works well for SNP chip data, but it may have statistical convergence issues in datasets with high marker density, such as those that result from resequencing studies of population samples. If you would like to phase that kind of dataset, please send a message to our mail list for suggestions about how to improve the quality of inference.
We have not yet posted instructions for how to reattach phased haplotypes across successive chunks along a chromosome. If you want to try this approach to phasing a whole chromosome, please send a message to our mail list.
Program Options
These links explain the command-line arguments that can be used to control IMPUTE2.
| Option type | Description |
|---|---|
| Required arguments | The program will not run if these are not supplied. |
| Input file options | A list of possible input files, with formatting requirements. |
| Output file options | Naming conventions and options for controlling format of output files. |
| Basic options | Options for controlling how the program processes input data. |
| Strand alignment options | Options for aligning allele coding across data files. |
| Filtering options | Options for controlling the filters that get applied to input data. |
| MCMC options | Options for controlling the MCMC algorithm. |
| Pre-phasing options | Options that facilitate pre-phasing and subsequent imputation. |
| Panel merging options | Options for merging a pair of reference panels. |
| Chromosome X options | Options for analyzing chromosome X data. |
| Expert options | Options to be used by experts only. |
Required arguments
This table shows the input arguments that you must supply in order for IMPUTE2 to run. These are just the minimum requirements; the program will not do anything useful unless you also supply other input options and/or data files.
| Flag | Default | Description |
|---|---|---|
| -g <file> REQUIRED unless |
none | File containing genotypes for a study cohort that you
want to impute or phase. The format of this file is
described on our file
format webpage and is the same as the output format
from our genotype calling program CHIAMO.
If you do not supply a file of unphased genotypes via this argument, you must supply a file of phased study haplotypes via the |
| -m <file> REQUIRED |
none | Fine-scale recombination map for the region to be
analyzed. This file should have three columns: physical
position (in base pairs), recombination rate between
current position and next position in map (in cM/Mb), and
genetic map position (in cM). The file should also have a
header line with an unbroken character string for each
column (e.g., "position COMBINED_rate(cM/Mb) Genetic_Map(cM)").
All of our |
| -int
<lower> <upper> REQUIRED |
none | Genomic interval to use for inference, as specified by
<lower>
and <upper>
boundaries in base pair position. The boundaries can be
expressed either in long form (e.g., IMPUTE2 requires that you specify an analysis interval in order to prevent accidental whole-chromosome analyses. If you want to impute a region larger than 7 Mb (which is not generally recommended), you must activate the |
Input file options
This table explains the formatting requirements for input data files that can be supplied to IMPUTE2. Some of these files allow more than one ID per SNP, but the program identifies SNPs internally by their base pair positions (which means that duplicate SNPs at a single position can cause problems). In all of these files, it is important that SNPs appear in base pair position order, from lowest to highest. It is also crucial that all SNP positions come from the same genome assembly (e.g., NCBI Build 37) so the program can combine information across input files.
| Flag | Default | Description |
|---|---|---|
| -g <file> REQUIRED unless |
none | File containing genotypes
for a study cohort that you want to impute or phase. The
format of this file is described on our file
format webpage and is the same as the output format
from our genotype calling program CHIAMO.
If you do not supply a file of unphased genotypes via this argument, you must supply a file of phased study haplotypes via the |
| -m <file> REQUIRED |
none | Fine-scale recombination map for the region to be
analyzed. This file should have three columns: physical
position (in base pairs), recombination rate between
current position and next position in map (in cM/Mb), and
genetic map position (in cM). The file should also have a
header line with an unbroken character string for each
column (e.g., "position COMBINED_rate(cM/Mb) Genetic_Map(cM)").
All of our |
| -h <file 1> <file 2> | none | File of known haplotypes, with one row per SNP and one
column per haplotype. All alleles must be coded as 0 or 1, and each In IMPUTE2, it is possible to specify two |
| -l <file 1> <file 2> | none | Legend file(s) with information about the SNPs in the -h file(s). Each
file should have four columns: rsID, physical position (in
base pairs), allele 0, and allele 1. The last two columns
specify the alleles underlying the 0/1 coding in the
corresponding -h
file; these alleles can take values in {A,C,G,T}. Each legend
file should also have a header line with an unbroken
character string for each column (e.g., "rsID position a0 a1").
We provide legend files for data from the HapMap Project
and the 1,000 Genomes Project in our When using two -h files with IMPUTE2, you must supply the corresponding legend files in the same order�i.e., the file with more SNPs comes first. |
| -g_ref <file> | none | File containing unphased genotypes to use as a reference panel for imputation. This file should follow the same format as the -g file. A -g_ref file can be used as the lone reference panel for imputation, or it can be combined with a single -h file to create a two-tiered reference panel (in the latter case, the -g_ref file should contain roughly a subset of the SNPs in the -h file). |
| -known_haps_g <file> | none | File containing known haplotypes for the study cohort.
The format is the same as the output format from IMPUTE2's
If your study dataset is fully phased, you can replace the The |
Output file options
The options in this table control the format and naming conventions of output files printed by IMPUTE2.
| Flag | Default | Description |
|---|---|---|
| |
|
Name of main output file. Follows the same format
as the |
| |
|
Name of SNP-wise information file with one line per SNP
and a single header line at the beginning. This file
always contains the following columns (header tags shown
in parentheses): 1. SNP identifier from 2. rsID (rs_id) 3. base pair position (position) 4. expected frequency of allele coded '1' in the -o file (exp_freq_a1) 5. measure of the observed statistical information associated with the allele frequency estimate (info) [details] 6. average certainty of best-guess genotypes (certainty) 7. internal "type" assigned to SNP (type) Depending on the command-line options invoked, there may also be columns labeled info_typeX, concord_typeX, and r2_typeX. IMPUTE2 assigns every SNP an internal "type" which reflects the combination of input datasets that include data for that SNP; here, X gives the type, which takes values in For SNPs that have genotypes in the -g file, concord_typeX is the concordance between the input genotypes and the best-guess imputed genotypes, where the input genotypes at that SNP have been masked internally and then imputed as if the SNP were of type X; similarly, r2_typeX is the squared correlation between input and masked/imputed genotypes at a SNP. The info_typeX column is the same information metric used in column 5, but here is it applied to genotypes that have been imputed from pseudo-type X SNPs in the leave-one-out masking experiment. These columns are useful for post-hoc quality control; we will soon explain how we use them in our section on Best Practices for Imputation. |
| |
|
Name of log file that records a summary of the screen output. |
| |
|
Name of file that records warnings generated by IMPUTE2. |
| |
|
"Output SNPs": specifies the SNP types that will be
printed to the output file (SNP labeling is discussed in
the Overview).
By default, all imputed and genotyped SNPs are included in
the output, i.e., " |
| |
|
Specifies that the main output file should be compressed by the gzip utility; this also applies to some non-standard output files that can become large. |
| |
3 | Specifies the number of decimal places to use for reporting genotype probabilities in the main output file. |
| |
|
Suppresses printing of info_typeX, concord_typeX, and r2_typeX columns in the -i file. |
| |
|
Suppresses printing of per-sample quality control
metrics file. The default is to print a file named " |
| |
|
IMPUTE2 always implicitly phases the study
genotypes ( In addition to this "best-guess" haplotype file, the program also prints the certainty that each successive pair of heterozygous SNPs is correctly phased. These certainties occur in a file named " As illustrated by our example commands, it is possible to use the |
| |
|
"Predict Genotyped SNPs": Tells the program to replace
the input genotypes from the |
| |
|
Unlike WARNING: This is an appealing option that will "fill in" sporadically missing genotypes in your input data. However, it is possible that this could cause subtle problems in downstream association testing. We therefore suggest that you use caution when applying this option. |
Details about 'info' metric
IMPUTE2 reports an information metric in the fifth
column of its
Our metric typically takes values between 0 and 1, where values near 1 indicate that a SNP has been imputed with high certainty. The metric can occasionally take negative values when the imputation is very uncertain, and we automatically assign a value of -1 when the metric is undefined (e.g., because it wasn't calculated).
Investigators often use the info metric to remove poorly imputed SNPs from their association testing results. There is no universal cutoff value for post-imputation SNP filtering; various groups have used cutoffs of 0.3 and 0.5, for example, but the right threshold for your analysis may differ. One way to assess different info thresholds is to see whether they produce sensible Q-Q plots, although we emphasize that Q-Q plots can look bad for many reasons besides your post-imputation filtering scheme.
We define our info metric and compare it against other metrics
in a
Basic options
These options control some basic processing that the program does to prepare input data for inference.
| Flag | Default | Description |
|---|---|---|
| REQUIRED |
none | Genomic interval to use for inference, as specified by
<lower>
and <upper>
boundaries in base pair position. The boundaries can be
expressed either in long form (e.g., IMPUTE2 requires that you specify an analysis interval in order to prevent accidental whole-chromosome analyses. If you want to impute a region larger than 7 Mb (which is not generally recommended), you must activate the |
| |
250 kb | Length of buffer region (in kb)
to include on each side of the analysis interval specified
by the -int option. SNPs
in the buffer regions inform the inference but do not
appear in output files (unless you activate the Using a buffer region helps prevent imputation quality from deteriorating near the edges of the analysis interval. Larger buffers may improve accuracy for low-frequency variants (since such variants tend to reside on long haplotype backgrounds) at the cost of longer running times. |
|
|
|
Allows the analysis of regions larger than 7 Mb. If this flag is not activated and the analysis interval plus buffer region exceeds 7 Mb, the program will quit with an error. The rationale for this flag is described here. |
|
|
|
Tells the program to include SNPs from the |
| |
20000 | "Effective size" of the population (commonly denoted as
Ne in the population genetics literature) from
which your dataset was sampled. This parameter scales the
recombination rates that IMPUTE2 uses to guide its
model of linkage disequilibrium patterns. When most
imputation runs were conducted with reference panels from
HapMap Phase 2, we suggested values of 11418 for imputation
from HapMap CEU, 17469
for YRI, and 14269
for CHB+JPT. Modern imputation analyses typically involve reference panels with greater ancestral diversity, which can make it hard to determine the "ideal" |
| |
0.9 | Threshold for calling genotypes in the -g file. For each
individual at each SNP, the program will use the genotype
with the maximum probability if that probability exceeds
the threshold; otherwise, the genotype will be treated as
missing. NOTE: This threshold applies only to input genotypes. If you want to apply a calling threshold to IMPUTE2's output probabilities, you will have to do it yourself. However, it is usually not a good idea to treat imputation output this way; see the webpage of our association-testing software SNPTEST for better suggestions. |
| |
# of indiv in |
Number of individuals from the -g file to include
in the analysis. For example, to impute only the first
five individuals, set |
| |
|
Print detailed output about the progress of imputation. By default, IMPUTE2 prints only the number of the current MCMC iteration when performing imputation, but this flag tells it to print more detailed updates. |
Strand alignment options
In any imputation analysis, is it absolutely essential that all panels have their allele codings aligned to a fixed reference (usually the human genome reference sequence). The options in this table are meant to help align the allele codings in your input data files, but you should not assume that the program will do all the work for you. If you do not know exactly how your data were processed or what these options are doing, you should try to locate the original strand information or send a message to our mail list for assistance.
NOTE: IMPUTE2 will automatically align the strand between panels whenever it can do so unambiguously; e.g., flipping A/C in Panel 2 to match G/T in the reference. The options below pertain to variants where this is not possible, e.g. because an A/T SNP cannot be aligned by label alone.
NOTE: We currently assume that all phased reference
files have already been aligned to the '+' strand of the human
genome reference sequence, which is true of
| Flag | Default | Description |
|---|---|---|
| |
none | File showing the strand orientation of the SNP allele
codings in the The ordering of the SNPs in this file does not matter (by contrast to the -g file, which must be sorted by SNP position), and it is okay if some SNPs in the strand file are not present in the genotype file (e.g., due to filtering). We provide model strand files in the Example/ directory that comes with the software download. |
| |
none | Same as |
| |
|
Activates the program's internal strand alignment
procedure for the NOTE: This flag can be used in conjunction with the -strand_g option. In that case, the information from the strand file takes precedence, i.e., the program will not try to align the strand of SNPs that have explicit strand info already. This is useful if you have strand information for some SNPs but not others. NOTE: You should take care when using this option. In particular, it can get the alignment wrong at A/T and C/G SNPs with minor allele frequencies near 50%, which can hurt the inference by distorting the local haplotype patterns. The best way to get the correct alignment at these kinds of SNPs is to track down the original assay and determine which strand was measured. This flag replaces -fix_strand_g as of IMPUTE v2.2. |
|
|
|
Similar to NOTE: Just as -align_by_maf_g can be used in conjunction with -strand_g, this flag can be used in conjunction with the -strand_g_ref option. As before, the strand file takes precedence over aligning the strand by MAF. NOTE: As with -align_by_maf_g, you should be careful about using this option to align A/T and C/G SNPs with minor allele frequencies near 50%. This flag replaces -fix_strand_g_ref as of IMPUTE v2.2. |
Filtering options
The options in this table affect the way that the program filters the input data. Some of the options provide direct control over which samples and SNPs get included in the analysis, while others set rules for how the program should behave when faced with certain filtering choices. These options are designed to make filtering more flexible, so that it is easy to apply any desired set of filters to a single underlying genotype file.
Some of these options apply to the dataset as a whole while others apply only to specific panels. The flag name for each panel-specific option ends in the command-line symbol for the file on which it operates; e.g., to exclude SNPs from the -g file you should use -exclude_snps_g, and to exclude SNPs from the -g_ref file you should use -exclude_snps_g_ref.
| Flag | Default | Description |
|---|---|---|
| |
none | This option provides flexible variant filtering in the
reference panel via "filter rules", which are based on
annotation columns in a To filter variants based on the numeric annotation values in the It is very important that you enclose each filtering string in single quotes, as shown above. Otherwise, the command-line environment may interpret symbols like You can develop annotations yourself and add them to the For an illustration of using |
| |
none | List of SNPs to exclude from the |
|
|
none | Same as |
| |
|
Specifies that SNPs excluded from the study dataset via
the |
| |
none | List of reference-panel-only SNPs to impute. If you do
not want the program to impute all of the reference SNPs
in the region you are analyzing, you can use this list to
specify a subset of SNPs to impute; all other SNPs will be
ignored unless they have data in the This option does not have any effect on SNPs in the |
| |
none | File of sample IDs for the individuals in the NOTE: Currently, the only reason to provide a sample file is if you want to exclude some individuals via the |
| |
none | Same as |
| |
none | List of samples to exclude from the NOTE: Part of the IMPUTE2 algorithm involves pooling information across the individuals in your study dataset. Samples with systematically aberrant genotypes (due, e.g., to degraded assay DNA) can confuse this part of the model; you should take care to identify such samples ahead of time and exclude them either manually or with this option. |
|
|
none | Same as |
MCMC options
IMPUTE2 uses an MCMC algorithm to integrate over the space of possible phase reconstructions for observed genotypes. The options in this table control the algorithm.
| Flag | Default | Description |
|---|---|---|
| |
30 | Total number of MCMC iterations to perform, including
burn-in. Increasing the number of iterations may
improve accuracy slightly, although increasing |
| |
10 | Number of MCMC iterations to discard as burn-in. The
algorithm samples new haplotypes for unphased individuals
during each of the first |
| |
80 | Number of haplotypes (in the reference or study data)
to use as templates when phasing observed genotypes.
Increasing this value will lead to higher accuracy at the
cost of longer running times, which scale quadratically
with |
| |
500 | Number of reference
haplotypes to use as templates when imputing missing
genotypes. As a rule of thumb, you
should set If all of your reference haplotypes have similar ancestry to the subjects in your study, each haplotype is potentially useful for imputation, so the best accuracy can be achieved by setting Conversely, we now recommend running IMPUTE2 with large reference panels containing haplotypes of diverse ancestry. (For more details, see here.) In this context, our rule of thumb suggests setting As of software version 2.3.0, |
Pre-phasing options
You can greatly speed up your imputation through a process
called
| Flag | Default | Description |
|---|---|---|
| |
|
Tells IMPUTE2 to phase the genotypes in the |
| |
|
Tells IMPUTE2 to perform imputation with
pre-phased GWAS haplotypes, which must be supplied via a We now recommend using SHAPEIT for pre-phasing and IMPUTE2 for downstream imputation. |
Panel merging options
These options allow IMPUTE2 to efficiently combine two reference panels typed on partially overlapping sets of variants.
| Flag | Default | Description |
|---|---|---|
| |
|
Tells the program to combine information across two reference panels using the approach described here. |
| |
none | Activates |
| |
none | Activates |
NOTE: If you want IMPUTE2 to print a merged
reference panel with buffer regions included, you should use one
of the last two options together with the
NOTE: You can see an example run that uses
Chromosome X options
These options facilitate the analysis of genotype data from human chromosome X.
| Flag | Default | Description |
|---|---|---|
| |
|
Specifies that this is an analysis of chromosome X
data. This flag changes the model parameters by
automatically reducing the When using the |
| |
|
Specifies that the current dataset comes from a pseudoautosomal
region (PAR) of chromosome X, where both males and
females are diploid. When used together with |
Expert options
The options in this table are meant for experts only. Don't use them unless you know what you are doing!
| Flag | Default | Description |
|---|---|---|
| |
random | Initial seed for random number generator. The seed is set using the system clock unless it is manually overridden with this option. |
| |
|
Turns warnings off, so that the |
| |
|
Turns on the "hole-filling" function, which allows SNPs
that are typed in the |
| |
|
Prevents the program from discarding SNPs whose alleles cannot be aligned across panels. Such SNPs will be retained in the output, but they will not be used for inference. |
Best Practices for Imputation
IMPUTE2 includes a rich collection of functions for analyzing genetic datasets, but it is most commonly used to perform genotype imputation in genome-wide association studies. To help investigators perform this kind of analysis, we have condensed the information on this website into a list of current best practices.
Pre-imputation filtering of study genotypes
Before you perform an imputation run with your study genotypes, you should filter the data to remove low-quality variants and individuals, as these can degrade the accuracy of the final results. Standard GWAS quality control filters are usually sufficient to prepare a dataset for imputation. It may also help to add an imputation-based QC step to the filtering process; we will describe this approach in the near future.
Variant position matching across input files
When you provide IMPUTE2 with reference and study data, the program determines which variants are shared across datasets by looking at their positions on the chromosome (as opposed, say, to their rsIDs). If two or more variants have the same position�perhaps because one is a SNP and one is an overlapping INDEL�then these variants are matched across panels based on their allele labels.
It is important to note that genomic coordinates change every couple of years as the human genome reference sequence is updated, so a given SNP may have different positions in different datasets. In order to obtain high-quality results from IMPUTE2, you must make sure that the variant positions in your input files are mapped to the same coordinate system, or "assembly".
Genomic assemblies are typically identified by their NCBI build number (e.g., "b36" or "b37") or their UCSC version (e.g., "hg18" or "hg19"). Our reference data download section shows the assembly to which each reference panel is mapped. If your study genotypes come from a different assembly than your reference panel, you should map the positions in your data to the reference coordinate system by using a tool like the liftOver program from UCSC. If you need help with this step, please send a message to our mail list.
Strand alignment between study and reference data
It is absolutely essential to align your study genotypes to the same strand convention as the reference panel from which you are imputing. Variants that are aligned to different strands may have different alleles (e.g., A/G in one dataset and T/C in another) or the same alleles at disparate frequencies (e.g., A/T in two datasets, where the 'A' allele occurs at 5% frequency in one dataset and 95% frequency in the other), and either of these scenarios can decrease imputation quality.
Most publicly available reference panels are aligned to the '+' strand of the human genome reference sequence, so the goal is to align your genotypes to the same convention. The best way to do this is to obtain assay information from the vendor who provided your genotypes; once you have this information, you can align your genotypes either manually or with the options described here. If you cannot recover the strand alignment from the original assay, you can use other options that tell IMPUTE2 to make educated guesses.
Choosing a reference panel
Historically, most GWAS investigators have tried to choose reference panels that match the ancestry of their study samples. We have developed a different approach: first supply IMPUTE2 with a worldwide reference panel, then let the program decide which haplotypes to use for imputation. This strategy can increase accuracy at low-frequency variants, and it avoids difficult choices about which haplotypes to include in the reference set. We currently recommend this approach for imputing genotypes in any human population. You can read our paper on this strategy here, learn about practical ways of applying it here, and download state-of-the-art reference haplotypes here.
If you have collected a custom reference panel for your study population�say, exome-wide or genome-wide sequencing data�you can combine it with the 1,000 Genomes data to maximize accuracy and genomic coverage at the same time. To learn how IMPUTE2 does this, see here.
Genome-wide imputation
It can be complicated and computationally demanding to impute thousands of individuals across the entire genome. We provide a few mechanisms to help with this process:
- IMPUTE2 includes command-line parameters that can be used to split the genome into discrete chunks for parallel analysis on a computing cluster. These parameters allow flexible partitioning of the genome with minimal manipulation of input files. See here for suggestions on how to use this functionality.
- IMPUTE2 is an efficient imputation method, but it still requires substantial computing time to process the whole genome in a large number of individuals. We have recently developed an approach called "pre-phasing" that greatly reduces the computational burden of imputation while sacrificing only a little accuracy; you can read more about the approach here. We now recommend this as the standard way of performing genome-wide imputation, although we still prefer the original IMPUTE2 MCMC algorithm for maximizing accuracy in smaller regions.
- Sequence-based reference panels contain large numbers of
rare and low-frequency variants, which can drive up the
computational cost of imputation. When computing power is
limited, it may be desirable to remove some of these variants
(e.g., those with very low frequencies in the population of
interest) before running imputation. To facilitate this
process, we have added the
-filt_rules_l option, which can flexibly remove reference variants based on command-line input to an IMPUTE2 run. You can see an example application of this approach and some guidelines for using it here.
Post-imputation filtering
It is standard practice to perform additional filtering once a batch of imputation runs has completed, mainly to remove poorly imputed variants that might behave badly in association tests. We are currently preparing some recommendations for this process; we will post them on the website as soon as they are ready.
Association testing
We distribute a program called SNPTEST that contains a powerful suite of statistical tests for association between phenotypes and imputed genotypes. You can download the software and read more about its functions at the SNPTEST website.
Follow-up imputation of putative associations
Once you have performed genome-wide imputation and association testing, you may want to take a closer look at regions with interesting associations. To get the best possible results, we recommend re-imputing this subset of regions with more intensive program settings:
- In contrast to the
pre-phasing approach that we recommend for genome-wide imputation, we suggest using the standard IMPUTE2 MCMC algorithm for follow-up imputation. This method takes longer to run in each region, but it should lead to slightly higher accuracy (especially at low-frequency variants) and remain computationally feasible when run on a limited portion of the genome. - If time permits, the overall accuracy may be improved by
increasing the value of the
-k parameter. - If time permits, the accuracy at low-frequency variants may
be improved by increasing the size of the
-buffer region�say, from the default value of 250 kb to 1000 kb (1 Mb).
Once you have re-imputed each region of interest, you should perform the association tests again to obtain a high-resolution estimate of the association landscape.
Pre-Phasing GWAS
Improvements in sequencing and genotyping technologies have rapidly increased the amount of reference data that can be used to impute untyped SNPs in association studies. Larger reference panels improve the power and resolution of imputation-based association mapping, but they also increase the computational burden of imputation. To help offset this cost, we have developed an extension of the IMPUTE2 methodology.
The basic idea is to "pre-phase" your study genotypes to produce best-guess haplotypes, then impute into these estimated haplotypes in a separate program run. By contrast, the original IMPUTE2 method integrates over the unknown phase of your study data during the course of an imputation analysis. Pre-phasing leads to a small loss of accuracy since the estimation uncertainty in the study haplotypes is ignored, but this allows for very fast imputation. This speedup is especially important because modern reference collections (such as those from the 1,000 Genomes Project) are frequently updated and expanded, so that many investigators would benefit from "re-imputing" their datasets following each reference panel update. The pre-phasing step needs to be performed just once per study dataset, so re-imputing is computationally cheap.
For these reasons, we now recommend pre-phasing as the
standard approach for genotype imputation in genome-wide
association studies, with the original IMPUTE2 algorithm
reserved for maximizing accuracy in more targeted analyses.
Pre-phasing is implemented through three program options:
We recommend performing the pre-phasing step with an accurate phasing method called SHAPEIT2 (details here and here), then imputing into the estimated GWAS haplotypes with IMPUTE2.
If you use this functionality in your study, please remember
to cite
Analyzing Whole Chromosomes
In principle, it is possible to impute genotypes across an entire chromosome in a single run of IMPUTE2. However, we prefer to split each chromosome into smaller chunks for analysis, both because the program produces higher accuracy over short genomic regions and because imputing a chromosome in chunks is a good computational strategy: the chunks can be imputed in parallel on multiple computer processors, thereby decreasing the real computing time and limiting the amount of memory needed for each run.
We therefore recommend using the program on regions of
The -int parameter provides
an easy way to break a chromosome into smaller chunks for
analysis by IMPUTE2. For example, if we wanted to split
a chromosome into
Once you have split a chromosome into multiple chunks and imputed them separately, the IMPUTE2 output format makes it easy to synthesize your results into a single whole-chromosome file. On linux-based systems, you can simply type a command like this:
cat chr16_chunk1.impute2 chr16_chunk2.impute2 chr16_chunk3.impute2 > chr16_chunkAll.impute2
Here, "chr16_chunkX.impute2" is an output file for one chunk of chromosome 16, and "chr16_chunkAll.impute2" is a combined output file that contains results for the entire chromosome. (Note that chr16 would typically need to be split into more than three chunks to satisfy the approximation used by IMPUTE2.)
Merging Reference Panels
Problem statement
Modern genotyping and sequencing technologies are generating a variety of reference datasets that can be used for genotype imputation in association studies. Combining reference panels from different populations can often improve imputation accuracy (e.g., see Howie et al. 2011), but it is not clear how best to merge panels that are genotyped at different sets of variants.
Howie et al. 2009 proposed a solution for the special case where one reference panel contains a subset of the variants in another reference panel. We previously released a combined 1,000 Genomes + HapMap 3 panel that takes advantage of this framework, and it was also used in the WTCCC2 studies.
Many association studies are now using the latest 1,000 Genomes data to drive their genotype imputation, but they may also have sequenced additional individuals from the population being studied. It makes sense to combine these resources in order to use all available reference information, but in this case each reference panel will contain many variants that are not found in the other�that is, the "hierarchical" variant framework of Howie et al. 2009 no longer applies.
With this in mind, we have devised a new strategy for combining reference panels created by different sequencing or genotyping studies.
Our approach
There are many possible ways to merge two reference panels. We are exploring several of these options, but we decided to start with the simple approach depicted in the figure below. The top panel of this figure shows two reference panels and a GWAS cohort; you can think of the rows as individuals and the columns as positions along the genome. Each vertical line represents a genotyped variant in a given panel, and each reference panel includes variants that are not found in the other.
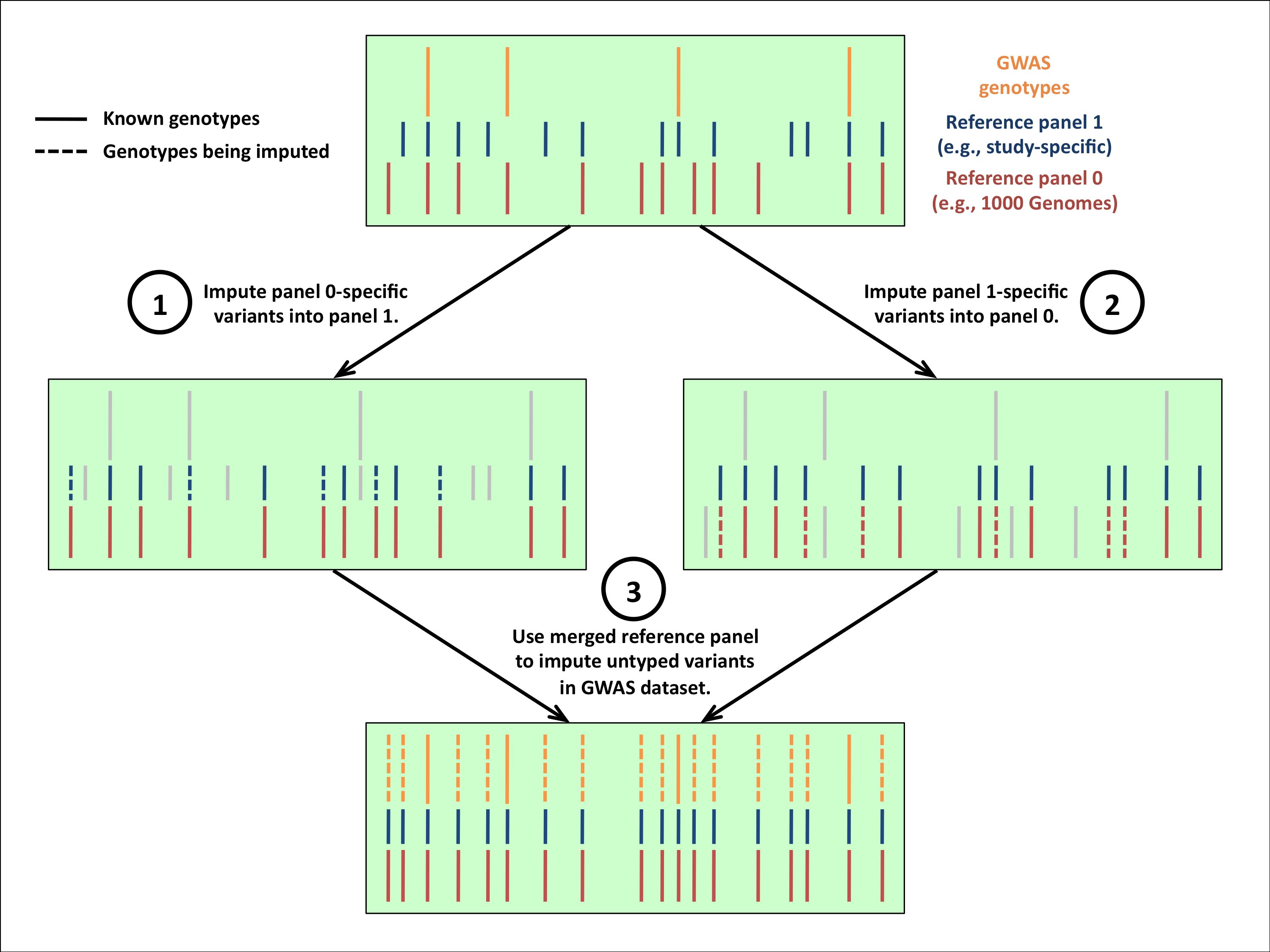
We impute the untyped variants in this figure in three steps:
- Impute the variants that are specific to Panel 0 (red) into Panel 1 (blue). Variants shown in grey do not inform the imputation.
- Impute the variants that are specific to Panel 1 (blue) into Panel 0 (red). Variants shown in grey do not inform the imputation.
- Now that we have imputed the two reference panels up to the union of their variants, treat the imputed haplotypes as known (i.e., take the best-guess haplotypes) and impute the GWAS cohort in the usual way.
This process can be performed with IMPUTE2 (version
2.3 and later) in a streamlined way: all you have to do is add
the
Practical considerations
Using pre-phased study data
The
Parameter settings
For finer control of the merging step, you can supply two
values to
Reference panel ordering
The order in which you supply the reference panels on the command line should not affect the accuracy of imputation from the merged panel: inside the program, the calculations are completely symmetric. One practical limitation is that only the first legend file in an IMPUTE2 command is allowed to have more than four columns. The 1,000 Genomes legend files we distribute typically have more than four columns, so if you are using these files it makes sense to provide the 1,000 Genomes panel before your other panel on the command line.
Printing the merged panel
By default, IMPUTE2 does not print the merged
reference panel (the outcome of Steps 1 and 2 above); the
merging is done internally, and the output shows only the
imputed genotypes for the study cohort. If you want the program
to output the merged panel, you can replace
-
-merge_ref_panels_output_ref �This option tells the software to merge the two reference panels and print the results in IMPUTE2 reference file format: one legend file and one haplotypes file. See the link for more information. -
-merge_ref_panels_output_gen �This option tells the software to merge the two reference panels and print the results in IMPUTE2 .gen file format. Phase information is ignored when creating this file, which can be useful if you want to re-phase the merged reference panel. See the link for more information.
If you want to merge two reference panels without imputing
into a study dataset (i.e., to skip Step 3 above), you
should use one of these two options and omit the study data (
Normally, these options print the merged reference panel
within the region specified by the
Publication and citation
Our approach for merging reference panels has not yet been published outside this website. We have tested the method on realistic datasets, and it has performed well in all of our analyses. We are actively working to document our work on this approach and to compare it with other strategies; we aim to report the results of these experiments and the details of our methodology as soon as possible.
In the meantime, we are happy to answer thoughtful questions
and to hear about your experiences with this new functionality.
If you would like to send comments, please do so through our
Imputation Concordance Tables
What is a concordance table?
Every run of IMPUTE2 produces a concordance table, except under certain settings that are not commonly used. A concordance table shows the results of an internal cross-validation that the program performs automatically. For this analysis, IMPUTE2 masks the genotypes of one variant at a time in the study data (Panel 2), then imputes the masked genotypes with information from the reference data and nearby study variants. The imputed genotypes are then compared with the original genotypes to evaluate the quality of the imputation. The results are summarized in a table like the one below:
If you are interested in the results of this experiment at a
given variant, you can find this information in the
How are concordance tables made?
Only variants with input data from a
The genotype probabilities from imputation are used somewhat
differently. In the first three columns of the table, we assign
each imputed genotype to a bin (
In the last three columns of the table, we again bin the
imputed genotypes based on their maximum posterior
probabilities, but this time the binning is cumulative: the bin
at the bottom of the table includes only genotypes that were
confidently imputed (max prob >= 0.9), while each bin above
includes all genotypes that pass a more lenient certainty
threshold. These thresholds are shown in the fourth column (
What can I do with a concordance table?
We can learn a couple of things from this kind of analysis.
First, the results can alert us to problems in the imputation:
if the concordance between imputed and input genotypes is
abnormally low, it may indicate that something went wrong in the
analysis or input files. A useful summary statistic is the
number in the upper righthand corner of the table, which gives
the overall concordance from the cross-validation. This number
should typically be around 95%; it may be lower in certain
populations or regions of the genome, but if it is much lower
then you may need to double-check the analysis. If you are
worried about your results, please send a message with details
of your analysis (including a
Concordance tables can also be used to predict the general quality of imputed genotypes at SNPs where we do not know the true genotypes. SNPs on GWAS microarrays tend to be easier to impute than untyped SNPs of the same frequency, so the cross-validation results may be somewhat optimistic, but they are often useful for relative comparisons�say, between different parameter settings of IMPUTE2.
Finally, the per-variant results of the cross-validation in
the output
Multiple reference panels
If you provide two reference panels to IMPUTE2, the program will perform the cross-validation in two different ways. First it will use only a single reference panel (Panel 0) to mimic Type 0 SNPs, and then it will use both reference panels together (Panels 0 and 1) to mimic Type 1 SNPs. In this case, IMPUTE2 will print two concordance tables�one for each type of reference SNP. Note that the same masked study genotypes are used to evaluate accuracy in both cases; the only difference is how much reference data we allow the program to see when imputing the masked genotypes.
Where can I find the concordance table?
The concordance table is printed at the end of an IMPUTE2
run. One copy is printed to STDOUT, and another copy is printed
in the
Scripts
The following scripts are designed to help with various parts of an IMPUTE2 analysis. We provide them in the hope that they will be useful, but we do not offer software support for them, and we cannot guarantee that they will work on your data due to inconsistencies in file formats, assumptions, etc. If you want to use one of these scripts, we suggest that you first read through the code to understand how it works.
All of these scripts are released under the GNU General Public License. Each script will print a list of command line options if you run it with no arguments.
| |
|
|---|---|
| vcf2impute_legend_haps.pl | Convert a phased VCF file into reference panel format: one legend file and one haplotypes file. |
| vcf2impute_gen.pl | Convert a phased or unphased VCF file into genotype file format (.gen). |
FAQ
Our FAQ has moved to this Google document.
References
[1] J. Marchini, B. Howie, S. Myers, G. McVean, and P. Donnelly (2007) A new multipoint method for genome-wide association studies via imputation of genotypes. Nature Genetics 39: 906-913 [Free Access PDF] [Supplementary Material] [News and Views Article]
[2] B. N. Howie, P. Donnelly, and J. Marchini (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genetics 5(6): e1000529 [Open Access Article] [Supplementary Material]
[3] J. Marchini and B. Howie (2010) Genotype imputation for genome-wide association studies. Nature Reviews Genetics 11: 499-511 [Restricted Access PDF] [Supplementary Material]
[4] B. Howie, J. Marchini, and M. Stephens (2011) Genotype imputation with thousands of genomes. G3: Genes, Genomics, Genetics 1(6): 457-470 [Open Access Article] [Supplementary Material]
[5] B. Howie, C. Fuchsberger, M. Stephens, J. Marchini, and G. R. Abecasis (2012) Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nature Genetics 44(8): 955-959 [Restricted Access PDF]
Contributors
The following people developed the methodology and software for IMPUTE2:
Bryan Howie, Jonathan Marchini
Mail List
If you have a question about IMPUTE2, please send a message to our mailing list:
http://www.jiscmail.ac.uk/OXSTATGEN
You will need to subscribe to the mailing list to post a question. The list has low but steady traffic, so you may want to redirect the messages to a dedicated e-mail folder if you don't want them all landing in your inbox.
IMPORTANT: If you are having a problem with the software, please include the following details in your e-mail; otherwise, we may not be able to diagnose the problem.
- The version number of IMPUTE2 and the type of
computer you are using to run it�e.g.,
"IMPUTE v2.2.2 on Mac OSX 10.6". - Any log files and/or screen output from the program; e.g., the "_summary" output file.
- For difficult problems like memory access errors (e.g., "segmentation faults"), we may need you to send data files that show the problem. These files should ideally be small, and we can provide suggestions if you are not allowed to share your actual data.