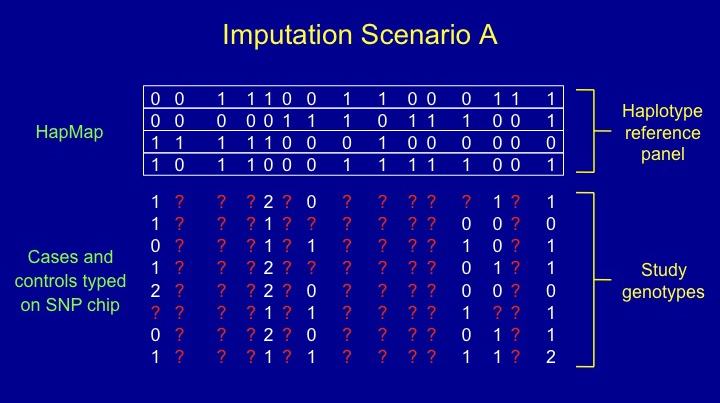
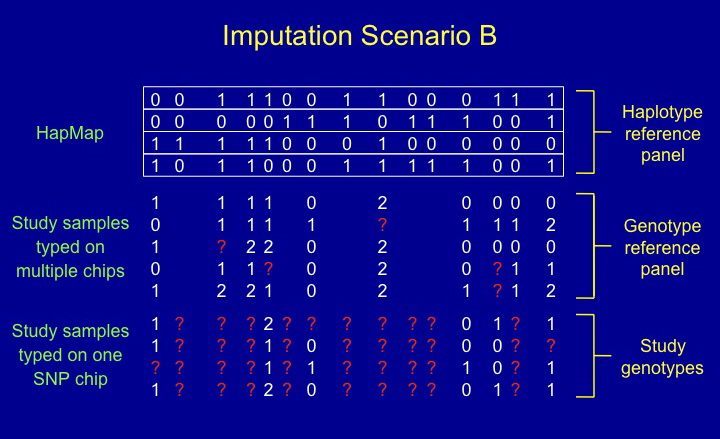
IMPUTE is a program for estimating ("imputing") unobserved genotypes in SNP association studies.
The program is designed to work seamlessly with the output of the genotype calling program
CHIAMO
and the population genetic simulator
HAPGEN,
and it produces output that can be analyzed using the program
SNPTEST.
There are currently three different versions of the IMPUTE software available for download:
version 0.5
implements the methodology described in
Marchini et al. (2007);
version 1
is essentially the same as version 0.5, with a couple of added features; and
version 2
implements a major extension that was introduced in
Howie et al. (2009). The situations
in which each version of the program can be applied are discussed below.
|
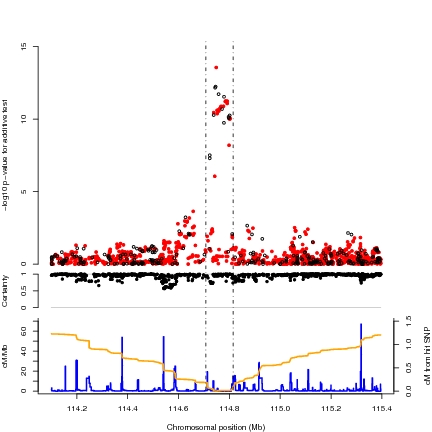
|