1,000 Genomes
haplotypes -- Phase I integrated variant set release (SHAPEIT2)
in NCBI build 37 (hg19) coordinates
NOTE : The haplotypes linked below
were released on 16 Jun 2014 and are an updated version of those
released on 9 Dec 2013. There was a small problem with the
strand of the Illumina OMNI data we used as the scaffold. 199
SNPs across the genome were not aligned to the + strand of the
human genome reference. This does not affect the phasing of the
haplotypes, but does affect downstream imputation, especially if
these SNPs were genotyped directly in the study being imputed.
A list of the 199 SNPs that have had their strand flipped is
here - 1000GP_16-06-14_flipped_SNPs.txt
These files are based on sequence data for 1,092 TGP samples from
ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20110521/
This dataset contains genotype likelihoods for 36,820,992 SNPs,
1,384,273 short bi-allelic indels and 14,017 structural variations
(SVs).
The haplotypes were phased using a new version of SHAPEIT2 that can
handle genotype likelihoods and genotypes available from SNP
microarrays on the same samples. The phasing proceeds in 2 steps
(i) Firstly the SNP array data are phased in order to build a
backbone (or 'scaff�old') of haplotypes across each chromosome.
(ii) We then use SHAPEIT2
to phase the sequence data 'onto' this haplotype sca�ffold.
This approach can take advantage of relatedness between sequenced
and non-sequenced samples to improve accuracy. The approach is
described in the following paper
Olivier Delaneau, Jonathan Marchini and the 1000 Genomes
Project Consortium (2013) Integrating
sequence and array data to create an improved 1000 Genomes
Project haplotype reference panel. Nature Communications
5:3934 doi:10.1038/ncomms4934
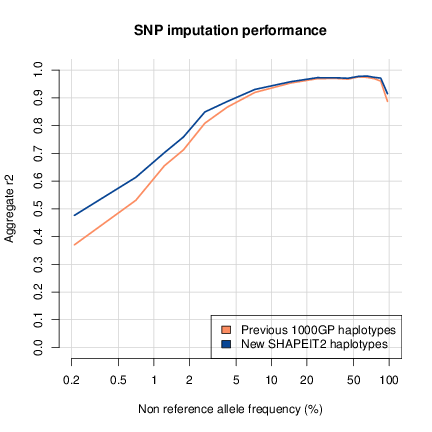
Using a set of validation genotypes at SNP and biallelic indels we
have been able to show that these haplotypes have lower genotype
discordance and improved imputation performance into downstream
GWAS samples, especially at low frequency variants. The following
figure shows the imputation performance of the previous 1000GP
haplotypes and this new release.
WARNING : these files are over 3Gb in size.
(i) ALL.integrated_phase1_SHAPEIT_16-06-14.nomono.tgz
- haplotypes at all polymorphic sites.
(ii) ALL.integrated_phase1_SHAPEIT_16-06-14.nosing.tgz
- haplotypes with singleton sites removed.
These files are gzipped tar archives and contain 4 kinds of files
:
1. *.haplotypes.gz
(one file per autosome) -- Phased haplotype file in
IMPUTE -h format (compressed by gzip software).
2. *.legend.gz
(one file per autosome) -- Legend file in IMPUTE -l format
(compressed by gzip software; variant positions in NCBI b37
coordinates). These files have the following columns
column 1 (id) - variant ID
column 2 (position) - base pair position
column 3 (a0) - allele labeled '0' in .hap
file. This is the reference allele.
column 4 (a1) - allele labeled '1' in .hap file
column 5 (type) - SNP/INDEL/SV denotes type of biallelic
variant
column 6 (source) - LOWCOV/EXOME denotes the type of sequencing used to generate the data at the
site
column 7-10 (afr.aaf amr.aaf asn.aaf eur.aaf) alternate
(non-reference) allele frequencies in 4 ancestral groups
column 11-14 (afr.maf amr.maf asn.maf eur.maf) minor
allele frequencies (MAF) in 4 ancestral groups
Filtering using the legend file We provide a single
worldwide haplotype file per chromosome (rather than splitting the
files by population or group) since IMPUTE2 is designed to
work with cosmopolitan reference panels via its -k_hap parameter.
One way to reduce the computational cost of imputation is to
flexibly remove certain SNPs on the command line via the
-filt_rules_l option. For example, if you are imputing into a
European dataset and want to ignore reference variants that are
monomorphic in the 1,000 Genomes EUR data, you can include "
-filt_rules_l 'eur.maf==0' " in your IMPUTE2 command,
which will tell the software to ignore SNPs with values of zero in
the 'eur.maf' column of the legend file. For more details about
-k_hap and -filt_rules_l, please see the IMPUTE2 website (http://mathgen.stats.ox.ac.uk/impute/impute_v2.html).
3. genetic_map*
(one file per autosome) -- Genetic map file in IMPUTE -m format
(physical positions in NCBI b37 coordinates).
4. *.sample
-- Text file with sample, population, group, and sex IDs for the
individuals in the haplotype files. The group IDs correspond
roughly to the continents of sampling, with occasional exceptions.
The sex IDs are '1' for males and '2' for females; these are
useful when interpreting data from the non-pseudoautosomal region
('nonPAR') of chromosome X. There are two haplotypes per sample,
and the column order of haplotypes matches the row order of sample
IDs. The population IDs are defined below, with group IDs in
square brackets and sample counts in parentheses.
ASW [AFR] (61) - African Ancestry in Southwest US
CEU [EUR] (85) - Utah residents (CEPH) with Northern and Western
European ancestry
CHB [ASN] (97) - Han Chinese in Beijing, China
CHS [ASN] (100) - Han Chinese South
CLM [AMR] (60) - Colombian in Medellin, Colombia
FIN [EUR] (93) - Finnish from Finland
GBR [EUR] (89) - British from England and Scotland
IBS [EUR] (14) - Iberian populations in Spain
JPT [ASN] (89) - Japanese in Toyko, Japan
LWK [AFR] (97) - Luhya in Webuye, Kenya
MXL [AMR] (66) - Mexican Ancestry in Los Angeles, CA
PUR [AMR] (55) - Puerto Rican in Puerto Rico
TSI [EUR] (98) - Toscani in Italia
YRI [AFR] (88) - Yoruba in Ibadan, Nigeria
-----------
TOTAL [AFR=246, AMR=181, ASN=286, EUR=379] (1092)