IMPUTE v0.5 has now been superseded by IMPUTE v1.0,
although we are keeping the website and software available for posterity. The description of
IMPUTE v1 below is equally applicable to IMPUTE v0.5.
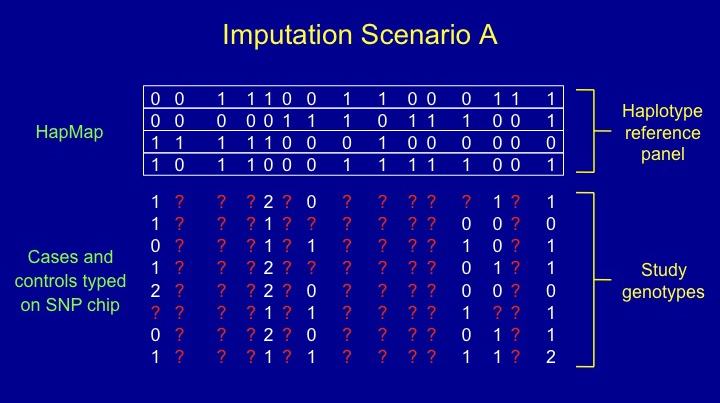
IMPUTE v1 (or "IMPUTE1", for short) is designed to
be used with a reference panel of known haplotypes,
such as those provided by the International
HapMap Project or the 1,000
Genomes Project, and a study sample genotyped at a subset of the
SNPs in the reference panel. IMPUTE1 fills in
missing genotypes (shown as red ?'s in the
figure above) by extrapolating linkage disequilibrium patterns from
the reference panel to the study individuals. This analysis scheme
is referred to as Scenario A by
Howie et al. (2009)
and in the figure above.
The basic method underlying IMPUTE1
(which was described by Marchini et al. [2007])
has been widely used to improve power in genome-wide association
studies. Until recently, the software implementing this method was
called IMPUTE v0.X.Y, where X
is an integer between 1 and 5. We are still supporting and developing
IMPUTE1.
The method is not ideal in all settings, however,
so we conducted a major revision
of the modeling framework and software; this revised approach is
implemented in IMPUTE v2, which is described
below.
IMPUTE v2 (or "IMPUTE2", for short) is based on the
same population genetic model as IMPUTE1,
but IMPUTE2 embeds this model in a more flexible
statistical framework. This framework allows IMPUTE2 to
increase accuracy (by using more of the information
in the data) and to handle a broader variety of imputation datasets.
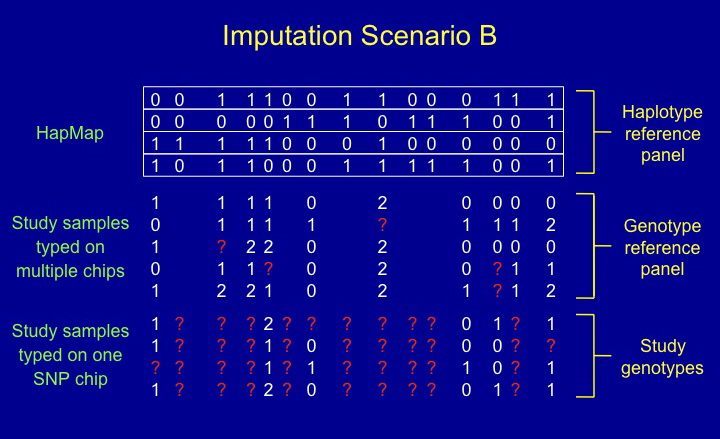
One important kind of dataset to which IMPUTE2 can be
applied is depicted above. In this example, we still want to
impute the missing genotypes in a set of study individuals (those in
the bottom panel), but we now have two different reference panels that
can inform the imputation: a set of known haplotypes (top panel), as
in Scenario A, and a set of unphased genotypes (middle panel)
observed at a subset of the SNPs in the top panel. This kind of
dataset, which is becoming increasingly common, is referred to as
Scenario B by Howie et al. (2009).
IMPUTE2 is uniquely suited to handle Scenario B in a
unified, integrated analysis framework. The
Howie et al. (2009) paper focused on the setting of
a case-control study where the controls form the middle panel and
the cases form the bottom panel, but the program can also be used
with other multi-tiered reference panels. Currently, it is most
commonly used with 1,000 Genomes haplotypes as the first reference
panel and HapMap 3 haplotypes as the second reference panel.
IMPUTE2 can also be applied in other imputation datasets that
pose problems for IMPUTE1,
including:
Datasets with a single, unphased reference panel (possibly with
sporadically missing genotypes)
Reference panels (phased or unphased) containing a large number
of chromosomes
Full details of IMPUTE2, including computational
considerations and accuracy comparisons
with other imputation programs, are provided in Howie et al. (2009).
We are still making a number of extensions and improvements to the
method, and we would appreciate any feedback that you might care to provide.
[1]
J. Marchini, B. Howie, S. Myers, G. McVean and P. Donnelly (2007)
A new multipoint method for genome-wide association studies via imputation of genotypes.
Nature Genetics 39: 906-913
[Free Access PDF]
[Supplementary Material]
[News and Views Article]
[2]
B. N. Howie, P. Donnelly and J. Marchini (2009)
A flexible and accurate genotype imputation method for the next generation of genome-wide association studies.
PLoS Genetics 5(6): e1000529
[Open Access Article]